![]()



cuBNM toolbox is designed for efficient brain network modeling on GPUs.
cuBNM toolbox uses GPUs to efficiently run simulations of brain network models consisting of nodes (neural mass models) which are connected through a connectome, and fit them to empirical neuroimaging data through integrated optimization algorithms.
GPU parallelization enables massive scaling of the simulations into higher number of simulations and nodes. Below you can see how computing time varies as a funciton of number of simulations and nodes on GPUs versus CPUs. For example, running 32,768 simulations (duration: 60s, nodes: 100) would take 4 days on a single CPU thread, but only 5 minutes on Nvidia A100 GPU, and 22 minutes on Nvidia GeForce RTX 4080 Super:
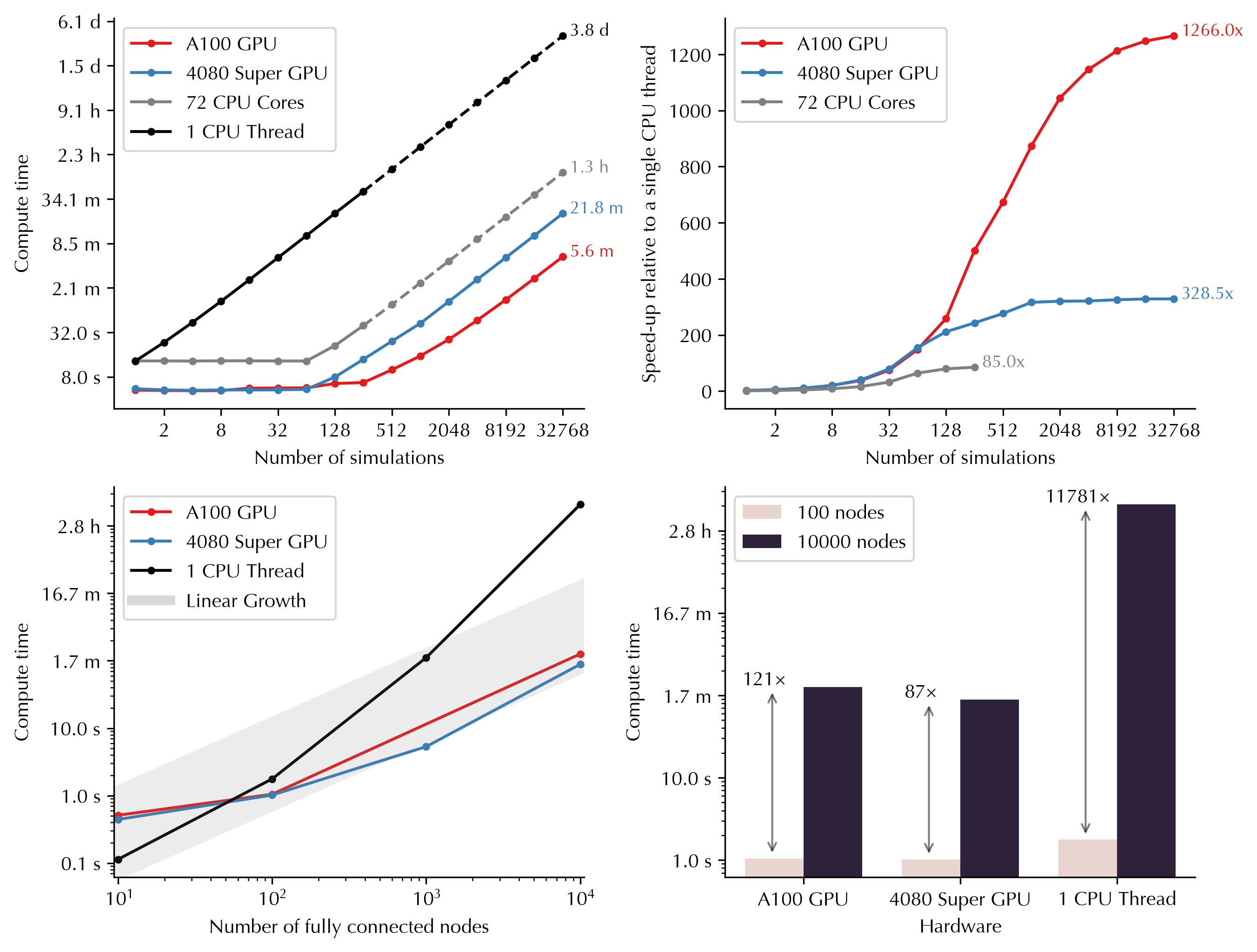
GPU usage is the primary focus of the toolbox but it also supports running the simulations on single or multiple cores of CPU. CPUs will be used if no GPUs are detected or if requested by the user.
Several commonly used models (e.g., reduced Wong-Wang, Jansen-Rit, Kuramoto, Wilson-Cowan) are implemented, and new models can be added via YAML definition files. A guide is included on the structure of model definition YAML fiels to help users implement their own models.
The simulated activity of model neurons is fed into the Balloon-Windkessel model
to calculate simulated BOLD signal. Functional connectivity (FC) and functional
connectivity dynamics (FCD) from the simulated BOLD signal are calculated efficiently
on GPUs/CPUs and compared to FC and FCD matrices derived from empirical BOLD signals
to assess similarity (goodness-of-fit) of the simulated to empirical BOLD signal.
The toolbox supports parameter optimization algorithms including grid search and
evolutionary optimizers (via pymoo), such as the covariance matrix adaptation-evolution
strategy (CMA-ES). Parallelization within the grid or the iterations of
evolutionary optimization is done at the level of simulations (across the GPU
‘blocks’), and nodes (across each block’s ‘threads’). The models can incorporate
global or regional free parameters that are fit to empirical data using the
provided optimization algorithms. Regional parameters can be homogeneous or vary
across nodes based on a parameterized combination of fixed maps or independent
free parameters for each node or group of nodes.
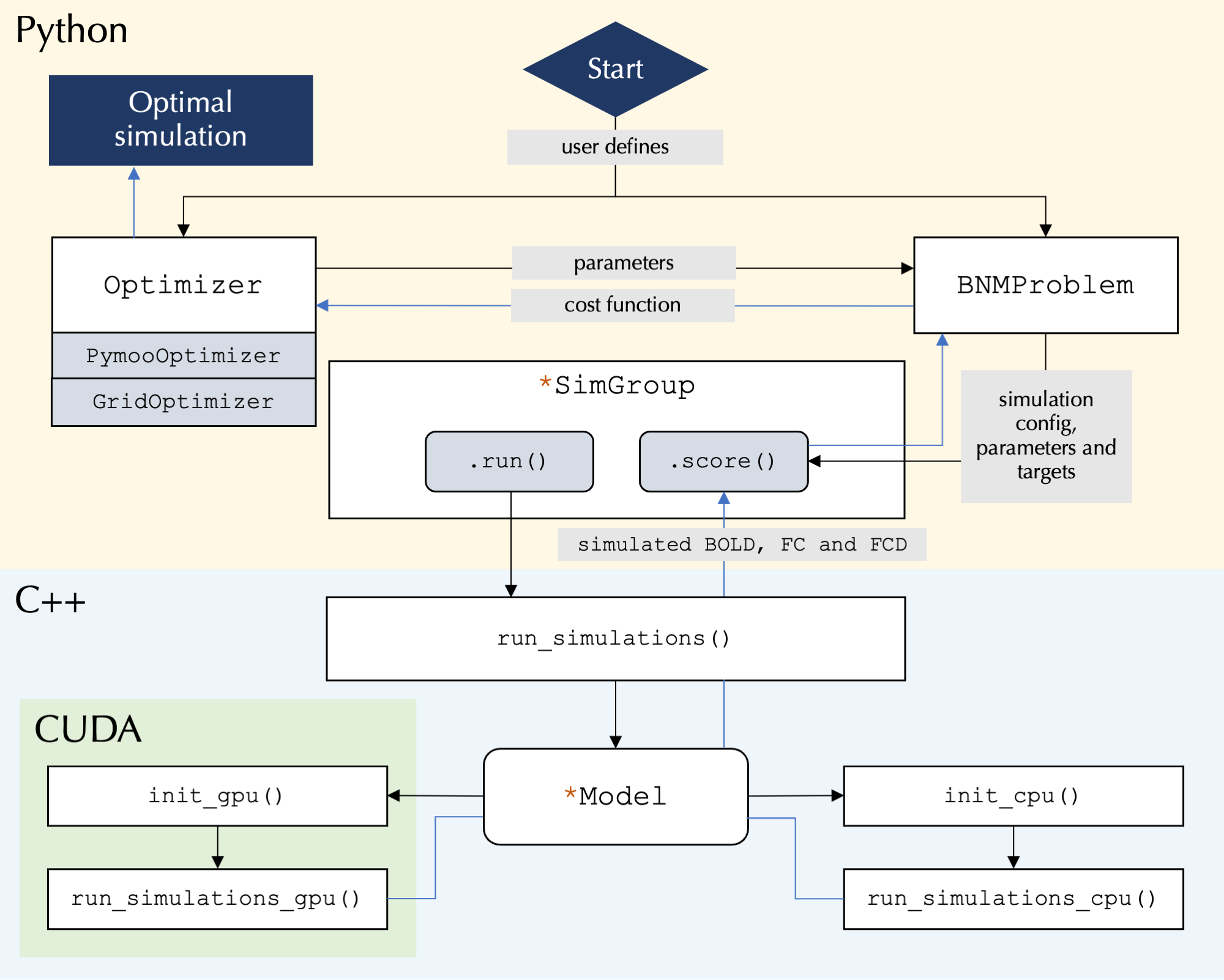
At its core the toolbox runs highly parallelized simulations using C++/CUDA, while the user interface is written in Python and allows for user control over simulation configurations:
Please find the documentations on installation, usage examples and API at https://cubnm.readthedocs.io.