Methods
Methodology of MicFunPred
MicFunPred relies on 32,453 genomes (permanent drafts/finished) downloaded from the Integrated Microbial Genome (IMG) database. Gene content tables were constructed in terms of Kegg Orthology (KO), Cluster Of Genes (COG), TIGRFAM, Enzyme Commission (EC), CAZymes and Protein Family (Pfam). A consolidated 16S rRNA gene database was developed using extracted sequences from genomes along with sequences from Greengenes (v13_5), SILVA (v132), and EZBiocloud (v2018.05) database. Sequences with length < 1200 and unannotated genera level taxonomy were filtered out and remaining sequences were clustered at 97% using Cd-hit to prepare the final database with 51,362 sequences.
MicFunPred predicts functional profiles from 16S rRNA gene amplicon sequence data by using a more conserved approach involving core genes of each genus, thereby minimizing the false-positive results. An abundance table and 16S rRNA gene sequences from OTU/ASV based approach can be used directly. First, OTU/ASV sequences are searched against a custom 16S rRNA gene database using BLAST. The taxonomy is assigned to each OTU/ASV at the genus level (if percent identity >= user cutoff) and the abundance table is then consolidated at assigned taxonomy level. The consolidated abundance table then normalized with mean 16S rRNA gene copy numbers for each taxon. The gene content (only core genes) for each genus listed in the abundance table is predicted. Core genes are defined as a set of all genes present in x% of genomes from each genus (the value of ‘x’ can be adjusted by the ‘Gene coverage’ cut-off). Using normalized abundance and predicted gene content table, metagenomes in terms of KO, EC, TIGRFAM, COG, CAZymes & Pfam are calculated by performing a dot product of two matrices. For pathway prediction, naive approaches can give inflated estimates of biological pathways. Hence, we have used MinPath, which gives estimates of a minimal set of pathways using a more conservative parsimony approach.
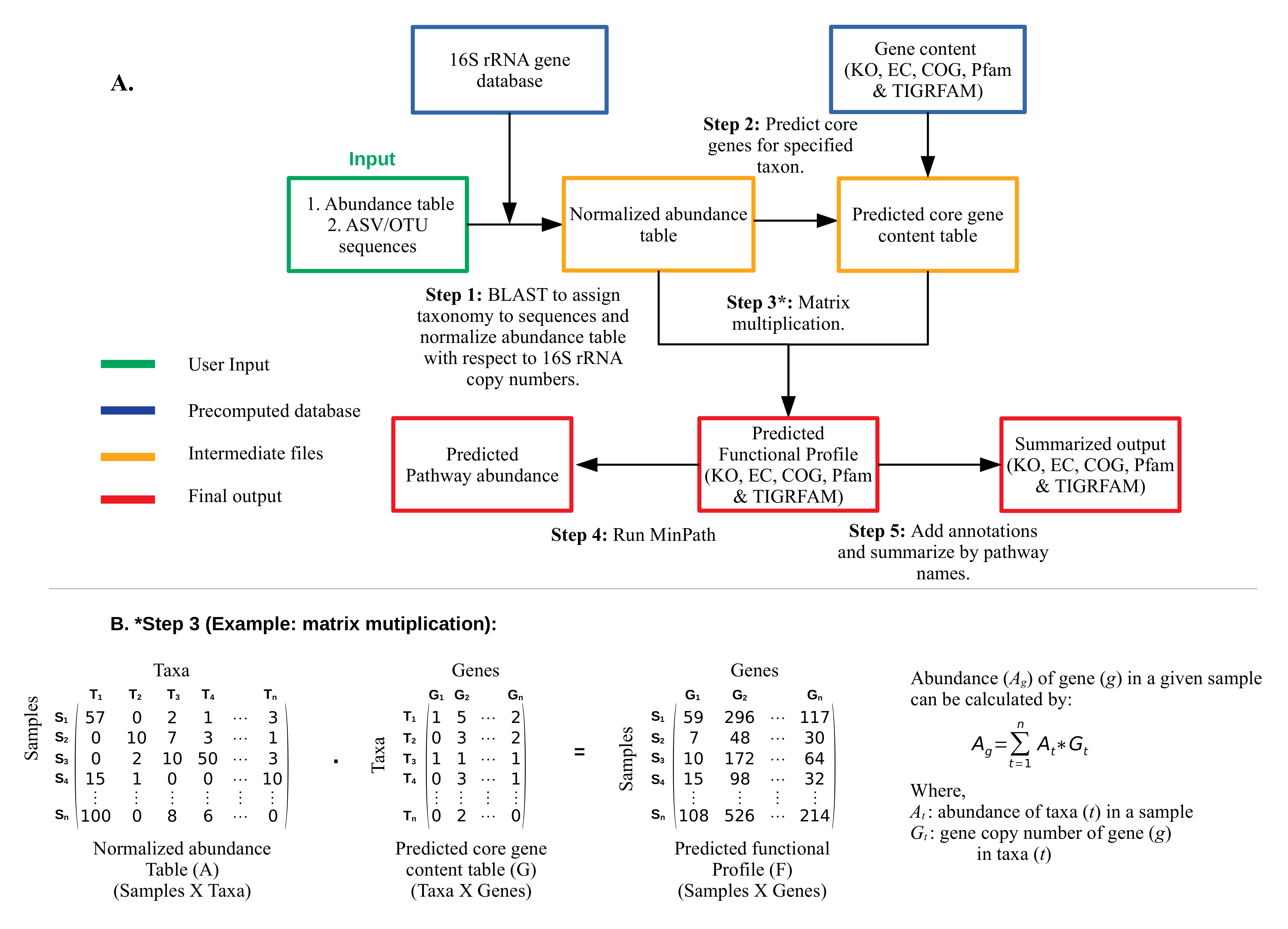
A) MicFunPred workflow: Abundance and 16S rRNA gene sequences from OTU/ASV based approach can be used with MicFunPred. The algorithm starts with comparing 16S sequences with a custom database, assignment of the genus (taxonomy), prediction of core genes of each assigned genera, and finally predicting metagenomic profiles by performing matrix-multiplication. In the last step, pathway predictions are made using MinPath for KEGG and MetaCyc annotations, while summarized abundances at different pathway levels are also generated. B) Matrix multiplication is explained using a hypothetical abundance and gene content table. The abundance of each gene in each sample is calculated by multiplying abundances with gene copy numbers and summing them together.