A Graphical User Interface (GUI) for prediction of SARS-CoV-2 spike protein binding energy with different ligands
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⢀⣀⣤⣤⣴⣶⡆⠀⣶⣶⣦⣤⣤⣄⠀ ⢠⣾⣿⣦
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣀⡀⢰⣿⣿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⡄⠘⢿⣿⠟
⠀⠀⠀⠀⠀⠀⠀⠀⢀⣴⣾⣿⣧⠀⢻⣿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⣿⠂⢠⣠⣤⣶⣦⡀
⠀⠀⠀⠀⠀⠀⣠⣾⣿⣿⣿⣿⣿⣇⠈⢿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⠏⢀⣿⣿⣿⣿⣿⣿⣦⡀
⠀⠀⠀⠀⠀⠘⢿⣿⣿⣿⣿⣿⣿⣿⡆⠘⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⡏⠀⣾⣿⣿⣿⣿⣿⣿⡿⠋⢀
⠀⠀⠀⣴⣿⣦⡀⠙⢿⣿⣿⣿⣿⣿⣿⡄⠸⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⡟⠀⣼⣿⣿⣿⣿⣿⣿⠟⢀⣴⣿⣧
⠀⠀⣼⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣿⣿⣷⡀⢹⣿⣿⣿⡇⠀⣿⣿⣿⡿⠁⣰⣿⣿⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣧
⠀⣼⣿⣿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣿⣧⠀⢻⣿⣿⡇⠀⣿⣿⣿⠃⢰⣿⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣿⣿⣿⣧
⠀⠛⠿⢿⣿⣿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣇⠀⢿⣿⡇⠀⣿⣿⠇⢠⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣿⣿⣿⣿⡿⠟⠃
⣼⣶⣤⣄⡈⠙⠛⠿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⡆⠈⠛⠃⠀⠛⠋⠀⣾⣿⠟⠁⣠⣾⣿⣿⣿⣿⡿⠿⠛⠉⣀⣠⣴⣶⡄
⣿⣿⣿⣿⣿⣿⣶⣤⣀⡉⠙⠻⢿⣿⣿⣦⠀⠁⠀⠀⠀⠀⠀⠀⠀⠀⠈⠁⢠⣾⣿⣿⠿⠛⠋⢁⣠⣴⣶⣿⣿⣿⣿⣿⣿⡀
⢻⣿⣿⣿⣿⣿⣿⣿⣿⣿⣷⣦⣤⣀⠉⠃⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠈⠋⢁⣀⣤⣶⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣧
⢸⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡏⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠘⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡟⠁⠀⣠⣶⣄
⠀⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⡄⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⡄⠀ ⢻⣿⣿⡿
⠀⠘⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠿⠓⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣸⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡿⠂⠀⠈⠙⠋
⠀⠀⠘⣿⣿⣿⣿⣿⠿⠛⠋⢁⣠⣤⣶⣧⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⢰⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠟⠋
⠀⠀⠀⠘⠟⠋⢉⣀⣤⣶⣿⣿⣿⣿⠟⠁⣠⣦⣄⡀⠀⠀⠀⠀⠀⠀⣤⣦⡈⠻⣿⣿⣿⣿⣿⣿⣿⣿⣿⡿⠛⠁
⠀⠀⠀⠀⠀⠘⢿⣿⣿⣿⣿⣿⠟⠁⣠⣾⣿⣿⣿⣿⣿⣶⣶⣾⣧⠀⢻⣿⣿⣦⡈⠻⣿⣿⣿⣿⡿⠟⠉
⠀⠀⠀⠀⠀⠀⠀⠙⠿⣿⡟⠁⣠⣾⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣇⠈⠿⢿⣿⣿⣦⡈⠻⠛⠉
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠈⠀⠺⢿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠋⢀⣤⣤⡈⠙⠋⠁
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠉⠙⠛⠻⠿⠿⠿⠿⠿⠿⠇⠀ ⣿⣿⣿⡇
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀ ⠈⠙⠋
/██ /██ /██ /██ /██████ /██████ /██████ /██ /██ /███████
| ██ | ██ | ███ | ██ |_ ██_/ /██__ ██ /██__ ██ | ███ /███ | ██__ ██
| ██ | ██ | ████| ██ | ██ | ██ \__/ | ██ \ ██ | ████ /████ | ██ \ ██
| ██ | ██ | ██ ██ ██ | ██ | ██ | ████████ | ██ ██/██ ██ | ███████/
| ██ | ██ | ██ ████ | ██ | ██ | ██__ ██ | ██ ███| ██ | ██____/
| ██ | ██ | ██\ ███ | ██ | ██ ██ | ██ | ██ | ██\ █ | ██ | ██
| ██████/ | ██ \ ██ /██████ | ██████/ | ██ | ██ | ██ \/ | ██ | ██
\______/ |__/ \__/ |______/ \______/ |__/ |__/ |__/ |__/ |__/
Version: 1.0.0 (in construction)
Important Note: The Excel table necessary for running the algorithm will be available only after the publication of the manuscript, as it contains data that, without a proper explanation (provided in the manuscript) can create false alarms about the containment of COVID-19. The data must be carefully and responsibly used, the information necessary for it is given in the manuscript.
Authors
Lilian Caroline Kramer Biasi
Opalina Vetrichelvan
Luís Fernando Mercier Franco
This material is supplementary to our manuscript "A Synergistic Approach of Molecular Docking and Artificial Intelligence for Drug Discovery: A Comprehensive Analysis of Ligand Structures Using SARS-CoV-2 as a Case Study (to pe published)".
Molecular docking simulations were carried out between two SARS-CoV-2 spike glycoprotein variants and more than 9,000 ligands. One of the spike variants was reported at the beginning of the pandemic (PDB: 6VSB), and the other contains mutation D614G and is fully cleaved by Furin (PDB: 7KDJ), which affects the affinity of the complex ligand-spike. From the molecular docking results of each SARS-CoV-2 spike protein variant mentioned above plus the results reported by de Oliveira et al. (2020), a machine learning model was trained, and can here be used for a fast prediction of the binding energy of different ligands with the spike variants.
Molecular docking, Machine Learning, Artificial Intelligence, SARS-CoV-2
- Graphical User Interface (GUI) for fast prediction of the binding energy of different ligands and SARS-CoV-2 spike proteins
- Artificial intelligence tool to accelerate drug design
- 1. Disclaimer
- 2. Language and Prerequisites
- 3. Building and Running
- 4. Citing us
- 5. Reporting Errors
The authors make no warranties about the use of this software. The authors hold no liabilities for the use of this software. The authors do not recommend the use of this software whatsoever. The algorithm is made freely available to clarify any details discussed in our manuscript. All information contained herein regarding any specific methodology does not constitute or imply its endorsement or recommendation by the authors.
The main program is written in Python 3.7, and use the following libraries that must be previously installed:
The code and the Excel file (containing the results of the molecular docking analyses) here available must be saved in the same folder.
Run the Covid-GUI.py file using a Python compiler of your preference. We used Spyder in Windows, no other compiler or operational system was tested.
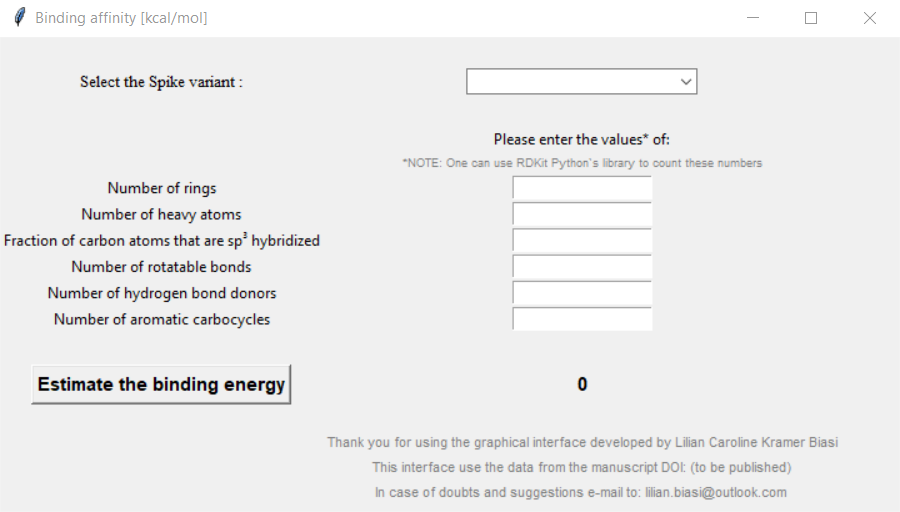
After running the code, an interface will open like this:
Figure 01: Program interface
Click here to zoom the figure in.
Select the spike protein variant:
Figure 02: Selecting the SARS-CoV-2 spike variant
Click here to zoom the figure in.
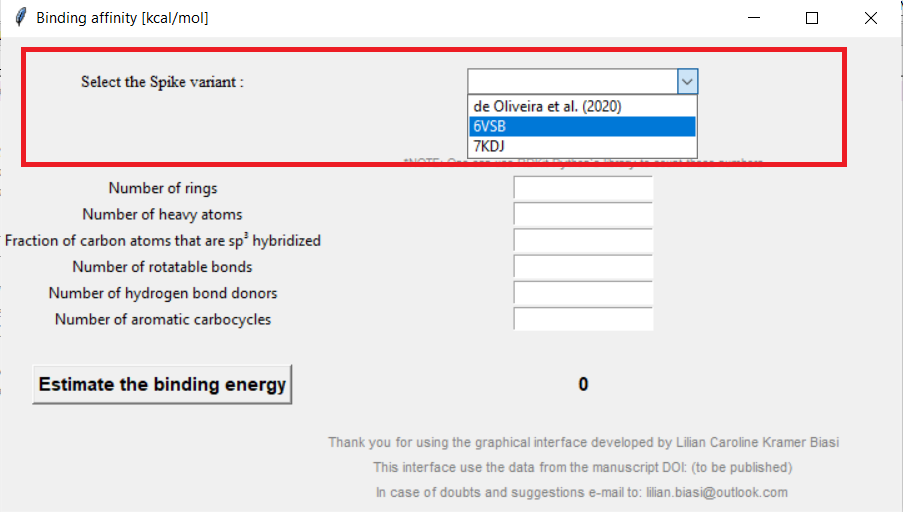
The available variants are the one elevated by de Oliveira et al. (2020), the one available as PDB: 6VSB, and the one available as PDB: 7KDJ. For more information about the variants, see our manuscript.
Enter the features of the ligand you desire to analyze, and then press the button to estimate the binding energy:
Figure 03: Entering the features (1) and calculating the binding energy (2)
Click here to zoom the figure in.
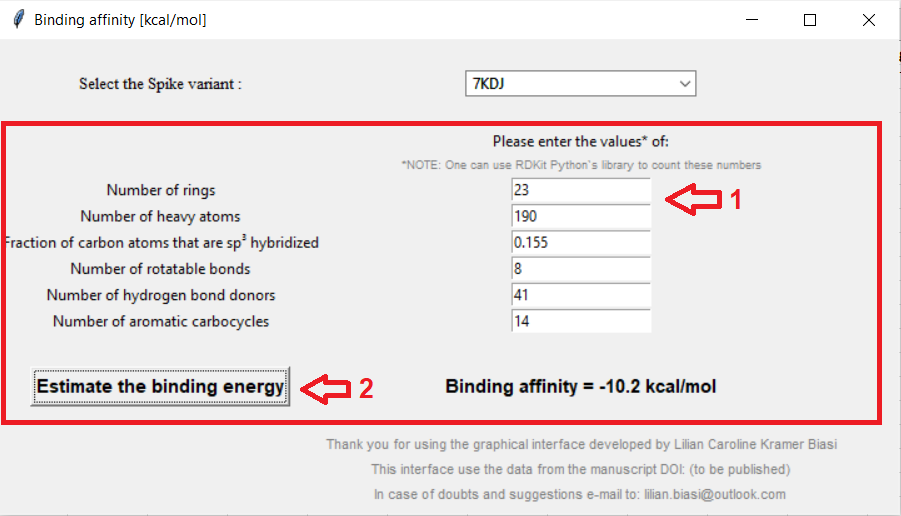
The required features can be manually counted from a molecule or can be counted using RDKit
If any of the codes here available was useful to you in any way, we kindly ask for you to cite our work: "Molecular docking coupled with machine learning to screen inhibitors of SARS-CoV-2: A comprehensive analysis of the structure of the potential ligands (to pe published)"
If you spot an error in the program files and all other documentation, please submit an issue report using the Issues tab.