![]()
-
2026.01.30: 发布 Gengram:检索增强的基因组基础模型,引入显式 motif memory、哈希加速的 k-mer 检索与更强的生物学可解释性。🚀 欢迎体验,看看它如何推动基因组建模。
-
2025.12.25: 新增竞品模型
NTv3-650M-Pre -
2025.12.18: Genos发布10B模型升级版本:Genos-10B-v2
- 训入新数据:引入非人灵长类与多种哺乳动物基因组
- 新增评测:(1)跨物种泛化能力;(2)超长上下文任务
- 推理优化:vLLM推理适配
- 多平台适配:华为昇腾,沐曦GPU适配
-
2025.10.23:Genos发布Genos-1.2B和Genos-10B
【模型架构与技术突破】
Genos作为人类基因组领域的基础模型,依托数百个高质量人类基因组基准数据进行训练,实现了对人类基因组序列长达百万碱基的上下文建模能力。通过单碱基级的分辨率学习,该模型具备了识别基因组中隐含的深层序列规律与功能特征的能力,为科学家构建起连接遗传信息与生命活动的新研究方法。本次发布包含12亿参数与100亿参数两个版本,均采用混合专家(MoE)架构,通过动态路由机制实现计算资源的优化配置,显著提升模型在复杂调控网络解析中的表现。
【功能模块与科学价值】
Genos具备精准识别关键功能元件的核心能力,能够深入解析微小基因变异对转录调控网络的级联效应,突破现在对非编码区调控元件的预测精度的传统方法局限,动态模拟变异位点对RNA表达谱的潜在影响,并追踪至表型形成的分子路径。在此基础上,研究团队开发了模块化应用接口,构建起"预测-解释-验证"的全链条研究体系。通过引入可解释性增强机制,该模型不仅提供高置信度的预测结果,更揭示调控网络中的关键节点与作用通路,为分子机制解析提供新的研究范式。
【开放生态与临床转化】
秉承开放科学理念,Genos在Github和Huggingface提供开源模型,并同时在DCS Cloud平台部署云端推理服务。研究者可下载模型进行部署及推理,或选择在DCS Cloud云端进行部署,我们还为使用者提供了从变异功能注释到表型预测的全流程分析示例代码,帮助使用者更快熟悉模型使用方法及功能。模型权重将进行持续更新,其在精准医学、群体健康、监测及发育生物学等领域的应用潜力将进一步释放。
【科学哲学与未来展望】
Genos为科学家研究基因的复杂调控及对功能的影响提供了新的可能性。未来随着跨模态学习能力的提升,Genos有望成为连接遗传密码与生命现象的"翻译器",在疾病预警、药物靶点发现及合成生物学等领域开启全新研究维度,目标实现从"基因组学"到"功能组学"的范式跨越。
- 人类核心语料:核心人类数据集由国际公认的联盟提供的单倍型解析和参考组装组成,包括来自人类泛基因组参考联盟(HPRC,V2)的 231 个组装,来自人类基因组结构变异联盟(HGSVC)的 65 个组装,来自人类多态性研究中心(CEPH)队列的 21 个基因组,以及 GRCh38 和 CHM13 参考基因组。在严格的质量控制后,该核心数据集包含 636 个高质量的人类基因组,总计约 2,443.5 亿碱基(对应于超过 1,500 亿个标记),并代表了多样化的全球人群。
- Genos-10B-v2 追加:BGI 的 CycloneSeq 平台生成的约 600 亿碱基的高覆盖率东亚人类基因组、来自 RefSeq 非人类灵长类基因组的 9501 亿碱基,以及来自 RefSeq 非灵长类哺乳动物基因组的 484.85 亿碱基,以 1:1 与核心人类语料分阶段混合。
- 质量控制:严格过滤与标准化,覆盖全球多样族群,确保单碱基精度与跨族群泛化。
- 基于 Transformer 的混合专家网络,Top-2 路由,25% FFN 稀疏。
- 超长上下文:RoPE 基数 50M,多维张量/管道/上下文/数据/专家并行;支持至 1M tokens。
- 训练稳定性:梯度裁剪、专家负载均衡(aux loss + z-loss)、GQA 50% KV 缓存压缩、Flash Attention。
- 推理:动态专家激活,单序列百万碱基推理可用。
模型参数:
| Genos-1.2B | Genos-10B | Genos-10B-v2 | |
|---|---|---|---|
| 总参数 | 1.2B | 10B | 10B |
| 激活参数 | 0.33B | 2.87B | 2.87B |
| 训练 tokens | 1600B | 2200B | 6286B |
| 架构 | MoE | MoE | MoE |
| 专家数 | 8 | 8 | 8 |
| Top-k | 2 | 2 | 2 |
| 层数 | 12 | 12 | 12 |
| Attention hidden | 1024 | 4096 | 4096 |
| 注意力头 | 16 | 16 | 16 |
| MoE FFN hidden | 4096 | 8192 | 8192 |
| 词表 | 128 (pad) | 256 (pad) | 256 (pad) |
| 最长上下文长度 | 1M | 1M | 1M |
- 更广物种覆盖:引入非人灵长类与多种哺乳动物序列,增强进化与保守性建模。
- 分阶段平衡混合:新增数据与人类核心语料 1:1 逐步混合,保持人类信号的主导性同时拓展多样性。
- 长程任务增强:在 DNALongBench 长程任务与跨物种分类上取得领先或并列领先表现。
- 长序列:DNALongBench(增强子-启动子关联、eQTL 等)。
- 短序列:基因元件/开放染色质/剪接位点等分类任务保持领先。
- 跨物种:多物种序列与基因元件分类任务验证 v2 的进化泛化能力。
- 变异热点与人群分类:在 8K/32K/128K 序列长度上稳定优于同类公开模型。
新增评测

初版评测

docker pull bgigenos/mega:v1
docker run -it --gpus all --shm-size 32g bgigenos/mega:v1 /bin/bash| 模型 | 总参数 | Hugging Face | Megatron ckpt |
|---|---|---|---|
| Genos-1.2B | 1.2B | Genos-1.2B | Genos-Megatron-1.2B |
| Genos-10B | 10B | Genos-10B | Genos-Megatron-10B |
| Genos-10B-v2 | 10B | Genos-10B-v2 | Genos-Megatron-10B-v2 |
pip install genos-client具体调用方式见 SDK 文档。
-
任务描述
本任务基于Genos大模型对于基因组的大量预训练积累,通过微调实现从DNA序列直接预测单碱基分辨率的RNA-seq表达谱,覆盖多种细胞类型和组织。其科学意义在于构建了基因组序列与转录组表达之间的直接映射关系,为理解基因调控机制和加速转录组学研究提供了创新工具
-
任务输入与输出
本任务属于回归任务。任务输入为hg38参考基因组部分序列(目前版本以32 kb为窗口),输出为不同细胞类型、基因组正负链对应序列位置的平均归一化RNA-seq信号值(单碱基精度)。核心是通过学习序列到表达的复杂映射,预测连续的转录组表达水平。
-
数据来源
训练数据来源于公共数据库ENCODE (ENCODE Consortium, 2012) 和GTEx (Kim-Hellmuth et al., 2020)。
数据经过整合后,共包含667个元数据组的单碱基转录组bigwig文件,同时输入hg38参考基因组。模型训练使用染色体1-22上的所有位置序列及其对应的平均RNA-seq谱作为配对数据。 目前公开版本使用了4例人B淋巴细胞和13例NK自然杀伤细胞进行微调,为去除样本间个体差异,两种细胞类型数据表达量取样本间平均表达量后均一化为各一组数据。
*目前公开版本已可实现其中人B淋巴细胞和NK自然杀伤细胞两种细胞类型1-22号染色体预测推理
-
模型设计
- 下游模型架构设计
模型在预训练的Genos-1.2B基础之上,替换原始输出头为任务专用的卷积模块。该模块由三层一维卷积层构成,通道维度逐层递减(1024→256→64→1),每层后接批归一化、GELU激活和丢弃正则化(dropout=0.1)。最终输出通过可学习权重参数缩放并经过Softplus激活,确保预测值为非负连续值,契合RNA-seq信号的回归特性。这种设计增强了模型对局部序列模式的捕捉能力,同时通过卷积的平移不变性优化了计算效率。
- 微调策略与训练优化
采用全参数微调(full fine-tuning),以均方误差(MSE)作为回归任务的损失函数。为解决RNA-seq信号值分布偏斜的问题,训练时引入平方根平滑裁剪和幂变换进行数值压缩,推理时执行逆操作以还原信号尺度。优化器选用Adafactor,配合余弦退火学习率调度器和线性预热(预热步数占总数5%),全局批量大小设为256,训练60个周期。这一策略在保证稳定收敛的同时,显著降低了长序列训练中的梯度波动风险。
- 基因组序列处理与上下文建模
为平衡长程依赖学习与计算成本,输入序列窗口长度设置为32 kb,相邻窗口重叠16 kb,覆盖染色体1-22的所有位点
(1). 指标及指标定义
评估核心指标为log1p变换后的皮尔逊相关系数(log1p-transformed Pearson correlation coefficient)。该指标用于衡量模型预测的RNA-seq谱与实验测得的真实谱在不同基因组范围内的一致性,具体计算范围包括:
-
全基因组(Whole genome):全基因组范围内,单碱基精度
-
基因表达量(Gene expression):基因表达量矩阵相关性,基因精度
*皮尔逊相关系数经过log1p(即log(1 + r))变换,以更好地评估信号预测的全局相关性。
(2). 测评指标
Genos模拟生成多细胞类型RNA-seq表达量与真实测序结果结果相关性0.9+。

(3). 输出示例

简介:
该项目为了验证多模态模型(基因模型+文本模型)在基因变异导致的疾病预测任务上,能够处理原始DNA序列,同时利用大语言模型的推理能力,生成具有生物学一致性的解释与预测。
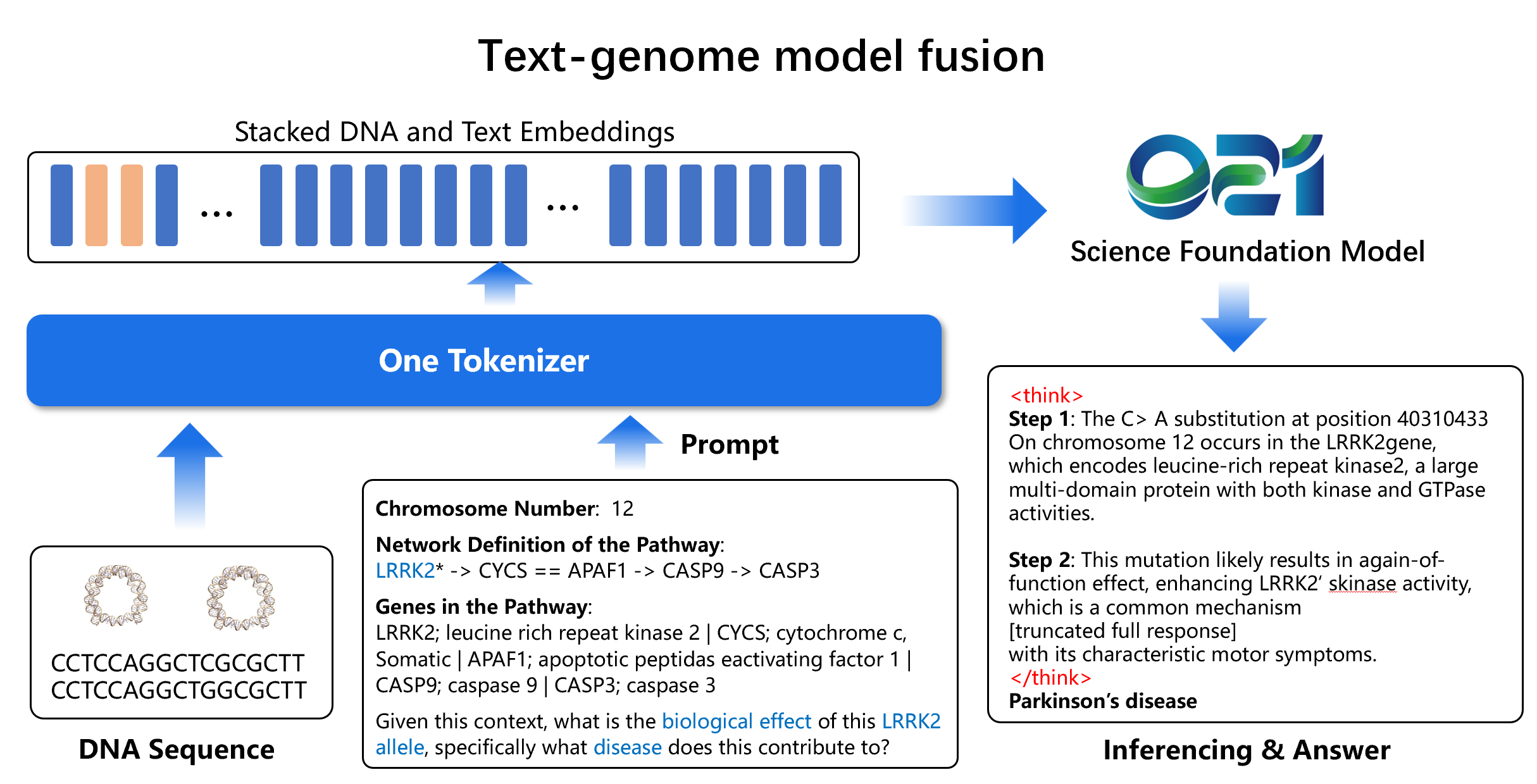
数据:
数据来源于论文Bioreason[7]当中的kegg任务,该任务数据通过多阶段整合KEGG通路与临床数据库变异信息,采用标准化符号系统表征分子网络中的各类相互作用,提供参考序列与变异序列的比。KEGG数据集包含1449条共计37种疾病信息,其中数据分配训练:验证:测试= 8:1:1。数据输入包括问题描述,参考基因序列与变异基因序列。输出包括推理和疾病分类预测。
模型设计:
-
架构设计:DNA模型、文本模型(Qwen3-1b、Qwen3-4b)、DNA embeding到文本embeding的投影层。
-
模型训练:本次模型训练旨在实现DNA序列与自然语言的高效对齐。训练当中冻结了基因模型(支持Evo2,Genos模型,),训练DNA embeding 到文本embeding的投影层,Lora微调文本模型,使用deepspeed策略进行高效训练。
-
基因序列处理:总共有两条基因序列:1、参考基因序列。2、变异基因序列。以变异基因为中心上下游,共计1024bp的基因序列。
评价指标:
- 准确率 (Accuracy): 正确预测的样本占总样本的比例。
不同模型在KEGG数据集上的结果对比情况如下表,其中基因模型中 Genos-10B 性能领先,文本-基因融合模型性能远超单独模态的模型,其中 021-8B与 Genos-1.2B融合模型准确率高达 98.28%,比单独用 Genos-1.2B高出 7%。

模型说明:
NT-2.5b-multi:InstaDeepAI/nucleotide-transformer-2.5b-multi-species
Evo2-1b:arcinstitute/evo2_1b_base
HyenaDna-1m: LongSafari/hyenadna-large-1m-seqlen
Genos-1.2B: BGI-HangzhouAI/Genos-1.2B
Genos-10B: BGI-HangzhouAI/Genos-10B
021-8B: 021 Science Foundation Model-8B is a large language model trained on extensive scientific corpora with profound scientific cognition. It is scheduled to be released at a later date.
-
vLLM 优化:我们在Genos上使用 vLLM 框架进行推理优化实验。该方案显著提高了吞吐量并减少了推理延迟。通过利用 vLLM 的创新 PagedAttention 算法和高效的内存管理机制,我们实现了与传统推理方法相比,吞吐量提高超过 7 倍。
- 拉取镜像
docker pull bgigenos/vllm:v1 docker run -it --entrypoint /bin/bash --gpus all --shm-size 32g bgigenos/vllm:v1
- 使用vllm进行embedding推理,请参考vllm example
-
其他硬件适配,请参考Adaptation
- 华为昇腾Ascend NPU
- 沐曦 GPU
- 训练数据均注明来源,核心人类语料来自 HPRC、HGSVC、CEPH、GRCh38、CHM13 等。
| 数据集 | 数据许可 | 来源 |
|---|---|---|
| HPRC Data Release 2 | MIT | 🌐 HPRC |
| HGSVC | 公开网站 | 🌐 HGSVC |
| CEPH | 公开网站 | 🌐 CEPH |
| GRCh38 | 公开网站 | 🌐 GRCh38 |
| CHM13 | 公开网站 | 🌐 CHM13 |
| High-coverage East Asian human genomes | 内部数据 | 邮件联系 |
| RefSeq non-human primate genomes | 公开网站 | 🌐 NCBI RefSeq |
| RefSeq non-primate mammalian genomes | 公开网站 | 🌐 NCBI RefSeq |
- 评测数据集整理中,将在huggingface项目主页持续更新。
- 模型与代码遵循 Apache License 2.0。
@article{10.1093/gigascience/giaf132,
author = {Genos Team, Hangzhou, China},
title = {Genos: A Human-Centric Genomic Foundation Model},
journal = {GigaScience},
pages = {giaf132},
year = {2025},
month = {10},
issn = {2047-217X},
doi = {10.1093/gigascience/giaf132},
url = {https://doi.org/10.1093/gigascience/giaf132},
eprint = {https://academic.oup.com/gigascience/advance-article-pdf/doi/10.1093/gigascience/giaf132/64848789/giaf132.pdf},
}
- 邮箱:Genos@genomics.cn
- 问题与建议:欢迎提交 Issue。
[1] The Human Genome Project. (2003). Finishing the euchromatic sequence of the human genome. Nature, 431(7011), 931 - 945.
[2] 1000 Genomes Project Consortium. (2010). A map of human genome variation from population - scale sequencing. Nature, 467(7319), 1061 - 1073.
[3] Grešová, K., Martinek, V., Čechák, D., Šimeček, P. & Alexiou, P. Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genomic Data 24, (2023).
[4] Dalla-Torre, H. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nature Methods (2024) doi:10.1038/s41592-024-02523-z.
[5] Trop, E. et al. The Genomics Long-Range Benchmark: Advancing DNA Language Models. OpenReview https://openreview.net/forum?id=8O9HLDrmtq.
[6] Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
[7] Fallahpour, Adibvafa, et al. BioReason: Incentivizing Multimodal Biological Reasoning within a DNA-LLM Model. arXiv preprint arXiv:2505.23579 (2025).
[8] Liao, W.W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).