These activities are designed for people with zero to some programming experience and zero to some Bioinformatics experience. We want to give Bioinformatics novices some of the confidence, skills, and resources needed to apply to labs/internships and to give non-Bioinformaticians a handle on how the field works. You guys are the trial group, since we have never approached UBIC classes this way. We ask for everyone to be patient and tell us when we are screwing up. Thanks!
In order to make sure everyone is on the same page at all times, we are going to have everyone use the same computer: a giant computer in the sky, EC2. We have an Ubuntu instance large enough to handle everyone's activities running in EC2, now we just need to have everyone make accounts. Please click here and enter your name and UCSD user name. If you cannot open the link, make sure you are logged into your UCSD email account. I will create an EC2 account for everyone - your username will be your UCSD username.
Secure Shell(ssh): a protocol which creates a secure channel for two computers to communicate even over an unsecured network. This is how we will connect to EC2.
How to prepare for ssh:
Windows: I do not like Windows terminals, you do not like Windows terminals, no one likes Windows terminals. Go to The Putty website and download Putty. A GUI should appear to guide you through the installation.
Mac or Linux: No preparation necessary, since you already have a native ssh client.
There are several very common and difficult problems in bioinformatics worth knowing, one of which is the problem of alignment. In order to understand why alignment is a problem, we need to undestand sequencing. You will have plenty of chances to memorize the steps of sequencing, and this is supposed to be about bioinformatics, so don't feel like you need to memorize them. I'll try to focus your attention on the problems that arise from sequencing (since that is where we, as bioinformaticists, step in).
Sanger Sequencing
This one is widely taught and known, but a little outdated.
1. Lyse the cells and extract DNA. The DNA itself is fragmented and copied many many times over.
2. Attach primers to the fragments and separate the experiment into 4 tubes.
3. Each tube receives plenty DNA polymerase, plenty deoxynucleosidetriphosphates (dNTPs) and about 1/100th the amount of di-deoxynucleotidetriphosphates (ddNTPs). The issue with ddNTPs is that they don't have the 3'-OH group. Synthesis stops when it a ddNTP is attached, which haappens at various points on various fragments.
4. DNA is negatively charged. Heat the DNA to separate it and put through a gel from pos->neg side, which separates fragments with a resolution of 1 nucleotide.
5. The trick is that the ddNTPs are flourescent! You now have many many fragments whose lengths you know down to the nucleotide. Expose an x-ray film to the gel, and now you have 4 rows representing the 4 nucleotides and dark bands where each of the 4 nucleotides terminated a fragment. Here's a picture to clarify:

Illumina Sequencing:
The most widely used method. This one is a bit harder to explain, so I recommend clicking here to watch a concise video on this topic.
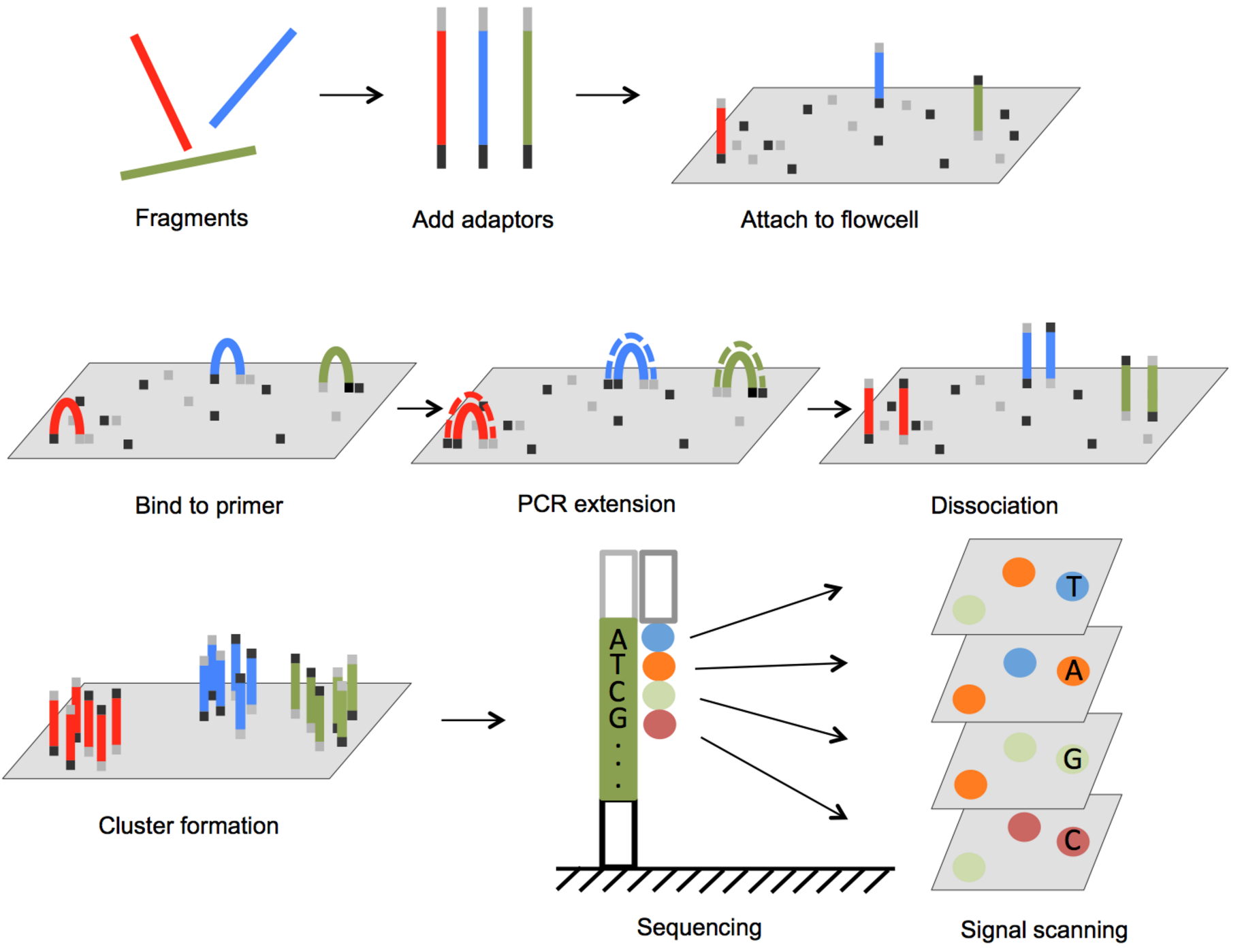
PacBio Sequencing:
1. DNA is immobolized at the bottom of a small well with a DNA polymerase.
2. Flourescent nucleotides are introduced to into the well, each labeled with a unique flourophore.
3. Light from the bottom of the well makes the bases fluoresce as they are added to the DNA template by the polymerase and the sequence of light colors is recorded.
So, what does it matter?
What do Sanger and Illumina sequencing have in common? Both produce ridiculous quantities of small DNA fragments. Illumina produces 300 million to 4 billion reads per run, with a selection of read lengths ranging from 50 base pairs to 300 base pairs. Meanwhile, Sanger produces 50000 sequences at lengths varying from 800 to 1000 base pairs. To give some perspective, the typical animal of interest is a human and those have 3.0×10^9 base pairs. Individual human genes range from 1148 to 37.7 kb (average length = 8446 bp,s.d. = 7124).
These tiny reads overlap all over the place. If you imagine the true sequence these reads came from and place the reads where they came from, you will get many reads piled up over every base pair in the true sequence. The more reads pile up, the more accurately you can predict the actual sequence. A common measure that rates the robustness of an alignment is coverage:

Alignment allows us to find how two or two thousand sequences line up, allowing us to identify single mutations, reduce error rates, build original(de novo) sequences, and analyze homology to build evolutionary trees.
Some problems to keep in mind for later
Each sequencing platform has its strengths and weaknesses that we will have to account for later.
Sanger
| Pros | Cons |
|---|---|
| - High precision - error rates of ~.001% | - low throughput |
| - long read length | - expensive |
Sanger sequencing is currently only really for small sequence lengths in a small number of samples.
Illumina
| Pros | Cons |
|---|---|
| - low cost | - short reads |
| - high throughput | - not cost effective for small number of targets |
| - decent precision - error rate: 0.46% - 2.4% |
Although the reads are short and the error rate is not as low as Sanger sequencing, Illumina sequencing produces so many sequences that it doesn't matter. Getting the most out of Illumina means getting the most out of its high throughput and per base coverage, which requires the proper software.
PacBio
| Pros | Cons |
|---|---|
| - long reads - up to 15kpb | - expensive |
| - high throughput | - high error rate |
Longer reads means that fewer reads are needed for the same coverage. Clearly, PacBio is best used for the reconstruction of longer genes or entire genomes.
Different tricks are available for different situations (Needleman-Wunsch for pairwise alignment or Burrows-Wheeler transform for aligning many reads to a single template), but we will be focusing mostly on how to use the tools rather than the algorithms within them.
TLDR: There are numerous complex applications of bioinformatics algorithms, from functional structure predictions to ancestral reconstructions. Alignment serves as the foundation for many of these algorithms, making basic sense of the incomprehensible mass of DNA that sequencing gives us.
The general idea behind clustering is the grouping of many elements so that the elements in a cluster are more similar to each other than they are to those in other groups.
Hierarchical Clustering:
Based on a system where elements that are closer together are more similar than those that are further apart. In this approach, elements which are close together will be combined into a cluster that is the hybrid location of both elements. So if we have an element a (0, 4) and another at (0, 2), the cluster is at (0, 3). The reason this is interesting to us is that it forms a tree of clusters, which can represent a tree of related genes which can be used to infer homology. Another example application is the finding of true sequences. Imagine you have a sequencing experiment that amplifies genes from an HIV sample, but you do not know how many successful mutations of HIV you have in the sample(infected people usually carry multiple versions because of how fast it mutates). After cleaning these sequences, clustering them together can reveal closely related groups. With the proper distance tweaking and statistical tricks, you can separate each real version of the virus into a cluster, revealing the prevalent mutations in the patient.

Side Note: As you will soon learn in your CSE classes, implementation is important and the state of the art alignment and clustering programs do their job quickly and accurately because they attempt to do the minimum amount of work possible. Fast alignment programs like mafft use fancy tricks like fourier tranforms and fast clustering algorithms often use simpler tricks like transforming into kmer representation. Bioinformatics has lots of data, so you should never attempt to solve a problem by going through all possible combinations or even the majority of all possible combinations. To give the classic stupid example, 80 sequences of length 1000 technically have over 1000^80 possible alignments which is a bit off from the 10^80 atoms in our universe.
Okay, we have had enough of conceptual stuff. Let's get at it with some cool visuals.
You will need java to proceed. If you do not have it, go here and press the big red install button in the middle of the page.
Aliview is a sequence viewer with a bunch of builtin tools, including alignment tools. We will use Aliview to see what a typical dataset looks like coming out of the illumina sequencing machine and what it means, visually, to align the sequences. Click here and go to download the stable version for your OS. Next, download a neat dataset I have for you from here. Launch aliview, click file->open file->PC64_V48_small_reconstructed_seqs.fasta. Scroll to the right and notice the mess that begins to form as you scroll. These sequences are sourced from the same gene and have gone through many steps, so the differences between them are quite likely to be real. Click align in the upper left corner and click realign everything. Now scroll forward and observe the gaps that have been inserted by the aligner.
These sequences are actually from HIV-1 glycoprotein envelopes from a person with broadly neutralizing antibodies against HIV-1(sequenced with PacBio). An effective HIV-1 vaccine should evoke the production of broadly neutralizing antibodies, so it is important to study the structure of the envelop that caused these antibodies to develop in individuals. The steps leading up to the data you downloaded allowed us to reveal a few specific strains. Now that we have them aligned, we can start asking questions about the differences between them and their evolution(which is important to figuring out how to counteract them). If you want to go into the nitty-gritty biology behind these sequences, go here. I will make a reconstruction of the evolutionary relationship for you to look at.
The SNP differences are obvious, but you will notice that there are weirder differences - like the >100 bp gaps formed in the middle. The truth is that I do not know why those are there! Those can be
- Real differences in hypervariable loops
- non-functional virions
- RT/PCR artifacts
Here's another cool tool: Blast will quickly look up a sequence in the NCBI database and spit out similar sequences it finds. Now, go to aliview and click edit->delete all gaps in all sequences. Copy the first sequence into the clipboard. Google NCBI Blast and open up the first result. Paste the sequence into the big box in the top of the page and click BLAST at the bottom. After a few seconds, Blast should link you to a whole lot of glycoprotein that are... from the article these were published in! This is one of the basic uses of Blast - to figure out where a sequence comes from.
In your email, you should have a password from me.
Windows: Open putty, paste ec2-18-191-97-249.us-east-2.compute.amazonaws.com into the Host Name section and select port 22 and SSH on that same page. Type a name under Saved Sessions and click the Save icon on the right. Now, press Open and type your username and password when prompted.
Mac or Linux: Right click anywhere and click open terminal. You should see a prompt that looks something like mchernys@mchernys-ThinkPad-T430:~/Desktop$. Next, copy paste this command into the terminal and press enter ssh your-username@ec2-18-191-77-154.us-east-2.compute.amazonaws.com. Note: use Ctrl-shift-V to paste into terminal. Please replace "your-username" with your actual username. Save the command you used somewhere so you can copy paste it in the future.
You probably have your own password in mind for your account. Type passwd username and follow the prompts to set your own password.
Discover your identity. Type whoami into the window that just opened up and hit enter. And just like that you're talking
with your computer, you bioinformatician, you.
Anytime you need a refresh on what a command does, type the command line with the --help option like so: ls --help. If that does not work, try man ls. I will go over why different commands have different help syntax in a bit.
In order to get started, we need to be able to do the same thing we do in a file explorer in the command line. You may find it inconvenient at first, but with time these commands become faster and more versatile than the file explorer's interface.
The forward slashes in a terminal console represent directories, with the home directory being a ~. Your default folder on EC2 is your use folder, which is ~/username. This means the folder named after your username is a subfolder of the home folder, which is represented by ~.
cd(change directory) Type cd followed by the directory's path to navigate a terminal to that directory. . is current directory and .. is the parent of the current directory.
ls(list files) prints out the contents of a directory. There are tons of options for this command - my favorite is ls -lah , since it prints the directory contents in list format(-l), includes hidden files/folders(-a), and makes the storage sizes more readable for humans(-h).
mkdir(make directory) Creates a directory with the same name as the argument you give it.
Navigate your terminal to your home directory(the directory named after your UCSD username) using cd. Type mkdir software and press enter. Type ls to see the changes you have made. The reason for a software folder is to keep your software in it, oddly enough. Usually, you would place executables in the /bin system folder, but you are not the admin so you cannot access that folder :( . This is often the case when you ssh into a system, so get used to having a dedicated software folder.