diff --git a/containers/2_ApplicationSpecific/GROMACS/EXAMPLES.md b/containers/2_ApplicationSpecific/GROMACS/EXAMPLES.md
new file mode 100644
index 00000000..ca71cfb0
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/EXAMPLES.md
@@ -0,0 +1,1967 @@
+# GROMACS Examples
+
+## Lysozyme in Water example
+
+Ref: http://www.mdtutorials.com/gmx/lysozyme/01_pdb2gmx.html
+
+
+Start an interactive Slurm job on a single node with a GPU, then change to your GROMACS directory e,g,
+
+```bash
+cd /projects/academic/[YourGroupName]/GROMACS
+```
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+Set environment variables to run the container
+
+```bash
+GROMACS_TAG="2023.2"
+container_image="gromacs-${GROMACS_TAG}-$(arch).sif"
+export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-$((${SLURM_JOB_CPUS_PER_NODE} / ${SLURM_GPUS_ON_NODE}))}"
+export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+mkdir -p "${CUDA_CACHE_PATH}"
+export GMX_ENABLE_DIRECT_GPU_COMM=1
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+```
+
+Fetch the input and data files for this exmaple from github:
+
+```bash
+if ! test -d "inputs"
+then
+ #git clone --depth 1 --single-branch --branch "GROMACS" https://github.com/tonykew/ccr-examples.git
+ git clone --depth 1 https://github.com/ubccr/ccr-examples.git
+ mv ./ccr-examples/containers/2_ApplicationSpecific/GROMACS/charmm36-jul2022.ff.tgz \
+ ./ccr-examples/containers/2_ApplicationSpecific/GROMACS/inputs .
+ rm -rf ccr-examples
+fi
+```
+
+Sample output:
+
+> ```bash
+> Cloning into 'ccr-examples'...
+> remote: Enumerating objects: 185, done.
+> remote: Counting objects: 100% (185/185), done.
+> remote: Compressing objects: 100% (155/155), done.
+> remote: Total 185 (delta 40), reused 115 (delta 24), pack-reused 0 (from 0)
+> Receiving objects: 100% (185/185), 1.34 MiB | 7.09 MiB/s, done.
+> Resolving deltas: 100% (40/40), done.
+> ```
+
+Start the container:
+
+```bash
+apptainer shell \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}"
+```
+
+Expected output:
+
+> ```bash
+> Apptainer>
+> ```
+
+
+All the commands from here, withing this terminal window, are run from the
+"Apptainer> " prompt
+
+Source the startup script to add "gmx" to the PATH
+
+```bash
+source /singularity
+```
+
+Sample output:
+
+> ```bash
+> CCRusername@cpn-c04-33:/projects/academic/[YourGroupName]/GROMACS$
+> ```
+
+
+This example uses the hen egg white lysozyme - PDB code 1AKI
+The PDB text file was downloaded from the [RCSB](http://www.rcsb.org/pdb/home/home.do) website for the crystal
+structure.
+
+This was downloaded as follows:
+
+Open a browser window to: https://www.rcsb.org
+Use the search bar top left to search for "1AKI"
+on the top left hand side of the window [Download Files] [Legacy PDB Format]
+download this file to a (new) "inputs" subdirectory
+
+Delete the crystal water molecules (residue "HOH" in the PDB file)
+
+```bash
+grep -v "HOH" "./inputs/1AKI.pdb" > "./inputs/1AKI_clean.pdb"
+```
+
+Verify thtat there a no entries listed under the comment MISSING
+Incomplete internal sequences or any amino acid residues that have missing
+atoms will cause pdb2gmx to fail
+
+```bash
+grep "MISSING" "./inputs/1AKI_clean.pdb"
+```
+
+No output expected
+
+
+This example uses the CHARMM36 force field, downloaded from the
+[MacKerell lab website](http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs)
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "charmm36-jul2022.ff.tgz" "https://mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/charmm36-jul2022.ff.tgz"
+tar xzvf "charmm36-jul2022.ff.tgz"
+```
+
+Use "gmx pdb2gmx" to generate three files:
+
+ The topology for the molecule.
+ A position restraint file.
+ A post-processed structure file.
+
+```bash
+gmx pdb2gmx -f ./inputs/1AKI_clean.pdb -o 1AKI_processed.gro -water tip3p
+```
+
+sample truncated output:
+
+> ```
+> :-) GROMACS - gmx pdb2gmx, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx_mpi
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx_mpi pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water tip3p
+>
+> Note that more recent versions of the CHARMM force field may be downloaded from
+> http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs.
+>
+> Select the Force Field:
+>
+> From current directory:
+>
+> 1: CHARMM all-atom force field
+>
+> From '/usr/local/gromacs/share/gromacs/top':
+>
+> 2: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
+>
+> 3: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
+> [...]
+> 15: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
+>
+> 16: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
+> ```
+
+Type "1" then [Enter] to select the "CHARMM all-atom force field" option
+
+```
+1
+```
+
+Sample truncated output:
+
+> ```
+>
+> Using the Charmm36-jul2022 force field in directory ./charmm36-jul2022.ff
+>
+> going to rename ./charmm36-jul2022.ff/aminoacids.r2b
+> Opening force field file ./charmm36-jul2022.ff/aminoacids.r2b
+> [...]
+> going to rename ./charmm36-jul2022.ff/solvent.r2b
+> Reading 1AKI_clean.pdb...
+> WARNING: all CONECT records are ignored
+> Read 'LYSOZYME', 1001 atoms
+>
+> Analyzing pdb file
+> Splitting chemical chains based on TER records or chain id changing.
+>
+> There are 1 chains and 0 blocks of water and 129 residues with 1001 atoms
+>
+> chain #res #atoms
+>
+> 1 'A' 129 1001
+>
+> All occupancies are one
+> All occupancies are one
+> Opening force field file ./charmm36-jul2022.ff/atomtypes.atp
+>
+> Reading residue database... (Charmm36-jul2022)
+> Opening force field file ./charmm36-jul2022.ff/aminoacids.rtp
+> [...]
+> Opening force field file ./charmm36-jul2022.ff/solvent.c.tdb
+> Analysing hydrogen-bonding network for automated assignment of histidine
+> protonation.
+> Processing chain 1 'A' (1001 atoms, 129 residues)
+> 213 donors and 184 acceptors were found.
+> There are 255 hydrogen bonds
+> Will use HISE for residue 15
+>
+> Identified residue LYS1 as a starting terminus.
+>
+> Identified residue LEU129 as a ending terminus.
+> 9 out of 9 lines of specbond.dat converted successfully
+> Special Atom Distance matrix:
+> CYS6 MET12 HIS15 CYS30 CYS64 CYS76 CYS80
+> SG48 SD87 NE2118 SG238 SG513 SG601 SG630
+> MET12 SD87 1.166
+> HIS15 NE2118 1.776 1.019
+> CYS30 SG238 1.406 1.054 2.069
+> CYS64 SG513 2.835 1.794 1.789 2.241
+> CYS76 SG601 2.704 1.551 1.468 2.116 0.765
+> CYS80 SG630 2.959 1.951 1.916 2.391 0.199 0.944
+> CYS94 SG724 2.550 1.407 1.382 1.975 0.665 0.202 0.855
+> MET105 SD799 1.827 0.911 1.683 0.888 1.849 1.461 2.036
+> CYS115 SG889 1.576 1.084 2.078 0.200 2.111 1.989 2.262
+> CYS127 SG981 0.197 1.072 1.721 1.313 2.799 2.622 2.934
+> CYS94 MET105 CYS115
+> SG724 SD799 SG889
+> MET105 SD799 1.381
+> CYS115 SG889 1.853 0.790
+> CYS127 SG981 2.475 1.686 1.483
+> Linking CYS-6 SG-48 and CYS-127 SG-981...
+> Linking CYS-30 SG-238 and CYS-115 SG-889...
+> Linking CYS-64 SG-513 and CYS-80 SG-630...
+> Linking CYS-76 SG-601 and CYS-94 SG-724...
+> Start terminus LYS-1: NH3+
+> End terminus LEU-129: COO-
+> Opening force field file ./charmm36-jul2022.ff/aminoacids.arn
+>
+> Checking for duplicate atoms....
+>
+> Generating any missing hydrogen atoms and/or adding termini.
+>
+> Now there are 129 residues with 1960 atoms
+>
+> Making bonds...
+>
+> Number of bonds was 1984, now 1984
+>
+> Generating angles, dihedrals and pairs...
+> Before cleaning: 5142 pairs
+> Before cleaning: 5187 dihedrals
+>
+> Making cmap torsions...
+>
+> There are 127 cmap torsion pairs
+>
+> There are 5187 dihedrals, 373 impropers, 3547 angles
+> 5106 pairs, 1984 bonds and 0 virtual sites
+>
+> Total mass 14313.255 a.m.u.
+>
+> Total charge 8.000 e
+>
+> Writing topology
+>
+> Writing coordinate file...
+>
+> --------- PLEASE NOTE ------------
+>
+> You have successfully generated a topology from: 1AKI_clean.pdb.
+>
+> The Charmm36-jul2022 force field and the tip3p water model are used.
+> [...]
+> ```
+
+This generates three files:
+
+```bash
+ls -l topol.top posre.itp 1AKI_processed.gro
+```
+
+Sample output:
+
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 88246 Nov 5 09:54 1AKI_processed.gro
+> -rw-rw-r-- 1 [CCRusername] nogroup 31304 Nov 5 09:54 posre.itp
+> -rw-rw-r-- 1 [CCRusername] nogroup 541409 Nov 5 09:54 topol.top
+> ```
+
+Define the box dimensions using the editconf module.
+
+```bash
+gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.2 -bt cubic
+```
+
+sample output:
+
+> ```
+> :-) GROMACS - gmx editconf, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.2 -bt cubic
+>
+> Note that major changes are planned in future for editconf, to improve usability and utility.
+> Read 1960 atoms
+> Volume: 123.376 nm^3, corresponds to roughly 55500 electrons
+> No velocities found
+> system size : 3.817 4.234 3.454 (nm)
+> diameter : 5.010 (nm)
+> center : 2.781 2.488 0.017 (nm)
+> box vectors : 5.906 6.845 3.052 (nm)
+> box angles : 90.00 90.00 90.00 (degrees)
+> box volume : 123.38 (nm^3)
+> shift : 0.924 1.217 3.688 (nm)
+> new center : 3.705 3.705 3.705 (nm)
+> new box vectors : 7.410 7.410 7.410 (nm)
+> new box angles : 90.00 90.00 90.00 (degrees)
+> new box volume : 406.88 (nm^3)
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l 1AKI_newbox.gro
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 88246 Nov 5 10:06 1AKI_newbox.gro
+> ```
+
+Fill the box with solvent (water) using the solvate module
+
+```bash
+gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
+```
+
+sample output:
+
+> ```
+> :-) GROMACS - gmx solvate, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
+>
+> Reading solute configuration
+> Reading solvent configuration
+>
+> Initialising inter-atomic distances...
+>
+> WARNING: Masses and atomic (Van der Waals) radii will be guessed
+> [...]
+> ++++ PLEASE READ AND CITE THE FOLLOWING REFERENCE ++++
+> A. Bondi
+> van der Waals Volumes and Radii
+> J. Phys. Chem. (1964)
+> DOI: 10.1021/j100785a001
+> -------- -------- --- Thank You --- -------- --------
+>
+> Generating solvent configuration
+> Will generate new solvent configuration of 4x4x4 boxes
+> Solvent box contains 41472 atoms in 13824 residues
+> Removed 1848 solvent atoms due to solvent-solvent overlap
+> Removed 1833 solvent atoms due to solute-solvent overlap
+> Sorting configuration
+> Found 1 molecule type:
+> SOL ( 3 atoms): 12597 residues
+> Generated solvent containing 37791 atoms in 12597 residues
+> Writing generated configuration to 1AKI_solv.gro
+>
+> Output configuration contains 39751 atoms in 12726 residues
+> Volume : 406.882 (nm^3)
+> Density : 988.485 (g/l)
+> Number of solvent molecules: 12597
+>
+> Processing topology
+> Adding line for 12597 solvent molecules with resname (SOL) to topology file (topol.top)
+>
+> Back Off! I just backed up topol.top to ./#topol.top.1#
+> [...]
+> ```
+
+This generates one file, "1AKI_solv.gro" and updates topol.top
+
+```bash
+ls -l 1AKI_solv.gro
+```
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 1788841 Nov 5 10:10 1AKI_solv.gro
+> ```
+
+```bash
+diff topol.top \#topol.top.1#
+```
+
+sample output:
+
+> ```
+> < LYSOZYME in water
+> ---
+> > LYSOZYME
+> 18486d18485
+> < SOL 12597
+> ```
+
+i.e. the "LYSOZYME" linewas changed to "LYSOZYME in water" and the
+"SOL [...]" line was added"
+
+
+
+Download the example molecular dynamics parameter (.mdp) file from
+http://www.mdtutorials.com/
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "./input/ions.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/ions.mdp"
+```
+
+Generate an atomic-level input file (.tpr)
+
+```bash
+gmx grompp -f inputs/ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
+```
+
+sample output:
+
+> ```
+> :-) GROMACS - gmx grompp, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx grompp -f inputs/ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
+>
+> Ignoring obsolete mdp entry 'ns_type'
+>
+> NOTE 1 [file inputs/ions.mdp]:
+> With Verlet lists the optimal nstlist is >= 10, with GPUs >= 20. Note
+> that with the Verlet scheme, nstlist has no effect on the accuracy of
+> your simulation.
+>
+> Setting the LD random seed to -102887427
+>
+> Generated 167799 of the 167910 non-bonded parameter combinations
+> Generating 1-4 interactions: fudge = 1
+>
+> Generated 117432 of the 167910 1-4 parameter combinations
+>
+> Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
+>
+> Excluding 2 bonded neighbours molecule type 'SOL'
+>
+> NOTE 2 [file topol.top, line 18486]:
+> System has non-zero total charge: 8.000000
+> Total charge should normally be an integer. See
+> https://manual.gromacs.org/current/user-guide/floating-point.html
+> for discussion on how close it should be to an integer.
+>
+>
+>
+> Analysing residue names:
+> There are: 129 Protein residues
+> There are: 12597 Water residues
+> Analysing Protein...
+> Number of degrees of freedom in T-Coupling group rest is 81459.00
+> The integrator does not provide a ensemble temperature, there is no system ensemble temperature
+>
+> NOTE 3 [file inputs/ions.mdp]:
+> You are using a plain Coulomb cut-off, which might produce artifacts.
+> You might want to consider using PME electrostatics.
+>
+>
+>
+> This run will generate roughly 3 Mb of data
+>
+> There were 3 NOTEs
+> [...]
+> ```
+
+This generates two files:
+
+```bash
+ls -l ions.tpr mdout.mdp
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 1173404 Nov 5 12:01 ions.tpr
+> -rw-rw-r-- 1 [CCRusername] nogroup 10972 Nov 5 12:01 mdout.mdp
+> ```
+
+Replace water molecules with the ions
+
+```bash
+gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
+```
+
+sample abridged output:
+
+```bash
+ :-) GROMACS - gmx genion, 2025.3 (-:
+
+Executable: /usr/local/gromacs/bin/gmx
+Data prefix: /usr/local/gromacs
+Working dir: /projects/academic/[YourGroupName]/GROMACS
+Command line:
+ gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
+
+Reading file ions.tpr, VERSION 2025.3 (single precision)
+Reading file ions.tpr, VERSION 2025.3 (single precision)
+Will try to add 0 NA ions and 8 CL ions.
+Select a continuous group of solvent molecules
+Group 0 ( System) has 39751 elements
+Group 1 ( Protein) has 1960 elements
+Group 2 ( Protein-H) has 1001 elements
+Group 3 ( C-alpha) has 129 elements
+Group 4 ( Backbone) has 387 elements
+Group 5 ( MainChain) has 515 elements
+Group 6 ( MainChain+Cb) has 632 elements
+Group 7 ( MainChain+H) has 644 elements
+Group 8 ( SideChain) has 1316 elements
+Group 9 ( SideChain-H) has 486 elements
+Group 10 ( Prot-Masses) has 1960 elements
+Group 11 ( non-Protein) has 37791 elements
+Group 12 ( Water) has 37791 elements
+Group 13 ( SOL) has 37791 elements
+Group 14 ( non-Water) has 1960 elements
+Select a group:
+```
+
+Select the "SOL" option "13"
+
+at the "Select a group: " prompt
+
+```bash
+13
+```
+
+sample output:
+
+> ```
+> Selected 13: 'SOL'
+> Number of (3-atomic) solvent molecules: 12597
+>
+> Processing topology
+> Replacing 8 solute molecules in topology file (topol.top) by 0 NA and 8 CL ions.
+>
+> Back Off! I just backed up topol.top to ./#topol.top.2#
+> Using random seed -209750018.
+> Replacing solvent molecule 7036 (atom 23068) with CL
+> Replacing solvent molecule 705 (atom 4075) with CL
+> Replacing solvent molecule 8703 (atom 28069) with CL
+> Replacing solvent molecule 12082 (atom 38206) with CL
+> Replacing solvent molecule 4139 (atom 14377) with CL
+> Replacing solvent molecule 12230 (atom 38650) with CL
+> Replacing solvent molecule 3250 (atom 11710) with CL
+> Replacing solvent molecule 10939 (atom 34777) with CL
+> [...]
+> ```
+
+This generates one file, "1AKI_solv_ions.gro" and updates topol.top
+
+```bash
+ls -l 1AKI_solv_ions.gro
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 1788130 Nov 5 12:13 1AKI_solv_ions.gro
+> ```
+
+```bash
+diff topol.top '#topol.top.2#'
+```
+
+sample output:
+
+
+> ```
+> < SOL 12589
+> < CL 8
+> ---
+> > SOL 12597
+> ```
+
+i.e. 8 water molecules have been replaced by CL ions
+
+Download the input parameter file "minim.mdp"
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "./input/minim.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/minim.mdp"
+```
+
+run the energy minimization
+
+```bash
+gmx grompp -f inputs/minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
+```
+
+sample output
+
+> ```
+> :-) GROMACS - gmx grompp, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx grompp -f inputs/minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
+>
+> Ignoring obsolete mdp entry 'ns_type'
+>
+> NOTE 1 [file inputs/minim.mdp]:
+> With Verlet lists the optimal nstlist is >= 10, with GPUs >= 20. Note
+> that with the Verlet scheme, nstlist has no effect on the accuracy of
+> your simulation.
+>
+> Setting the LD random seed to -67666049
+>
+> Generated 167799 of the 167910 non-bonded parameter combinations
+> Generating 1-4 interactions: fudge = 1
+>
+> Generated 117432 of the 167910 1-4 parameter combinations
+>
+> Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
+>
+> Excluding 2 bonded neighbours molecule type 'SOL'
+>
+> Excluding 3 bonded neighbours molecule type 'CL'
+> Analysing residue names:
+> There are: 129 Protein residues
+> There are: 12589 Water residues
+> There are: 8 Ion residues
+> Analysing Protein...
+> Number of degrees of freedom in T-Coupling group rest is 81435.00
+> The integrator does not provide a ensemble temperature, there is no system ensemble temperature
+>
+> The largest distance between excluded atoms is 0.443 nm between atom 1156 and 1405
+> Calculating fourier grid dimensions for X Y Z
+> Using a fourier grid of 64x64x64, spacing 0.116 0.116 0.116
+>
+> Estimate for the relative computational load of the PME mesh part: 0.32
+>
+> This run will generate roughly 3 Mb of data
+>
+> There was 1 NOTE
+> [...]
+> ```
+
+This generates one file, "em.tpr" and updates mdout.mdp
+
+```bash
+ls -l em.tpr mdout.mdp
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 1174552 Nov 6 10:26 em.tpr
+> -rw-rw-r-- 1 [CCRusername] nogroup 10614 Nov 6 10:26 mdout.mdp
+> ```
+
+
+Run the energy minimization
+
+```bash
+gmx mdrun -v -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm em
+```
+
+sample abridged output:
+
+> ```
+> :-) GROMACS - gmx mdrun, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx mdrun -v -ntmpi 2 -deffnm em
+>
+> Reading file em.tpr, VERSION 2025.3 (single precision)
+> Using 1 MPI thread
+> Using 40 OpenMP threads
+>
+>
+> Steepest Descents:
+> Tolerance (Fmax) = 1.00000e+03
+> Number of steps = 50000
+> Step= 0, Dmax= 1.0e-02 nm, Epot= -4.55681e+05 Fmax= 1.81034e+05, atom= 1891
+> Step= 1, Dmax= 1.0e-02 nm, Epot= -4.67710e+05 Fmax= 6.69947e+04, atom= 936
+> Step= 2, Dmax= 1.2e-02 nm, Epot= -4.81031e+05 Fmax= 2.92839e+04, atom= 19487
+> [...]
+> Step= 448, Dmax= 1.1e-02 nm, Epot= -6.22184e+05 Fmax= 8.05544e+03, atom= 567
+> Step= 449, Dmax= 1.3e-02 nm, Epot= -6.22194e+05 Fmax= 9.69387e+03, atom= 567
+> Step= 451, Dmax= 7.8e-03 nm, Epot= -6.22300e+05 Fmax= 9.55374e+02, atom= 567
+>
+> writing lowest energy coordinates.
+>
+> Steepest Descents converged to Fmax < 1000 in 452 steps
+> Potential Energy = -6.2229994e+05
+> Maximum force = 9.5537390e+02 on atom 567
+> Norm of force = 2.5618381e+01
+> [...]
+> ```
+
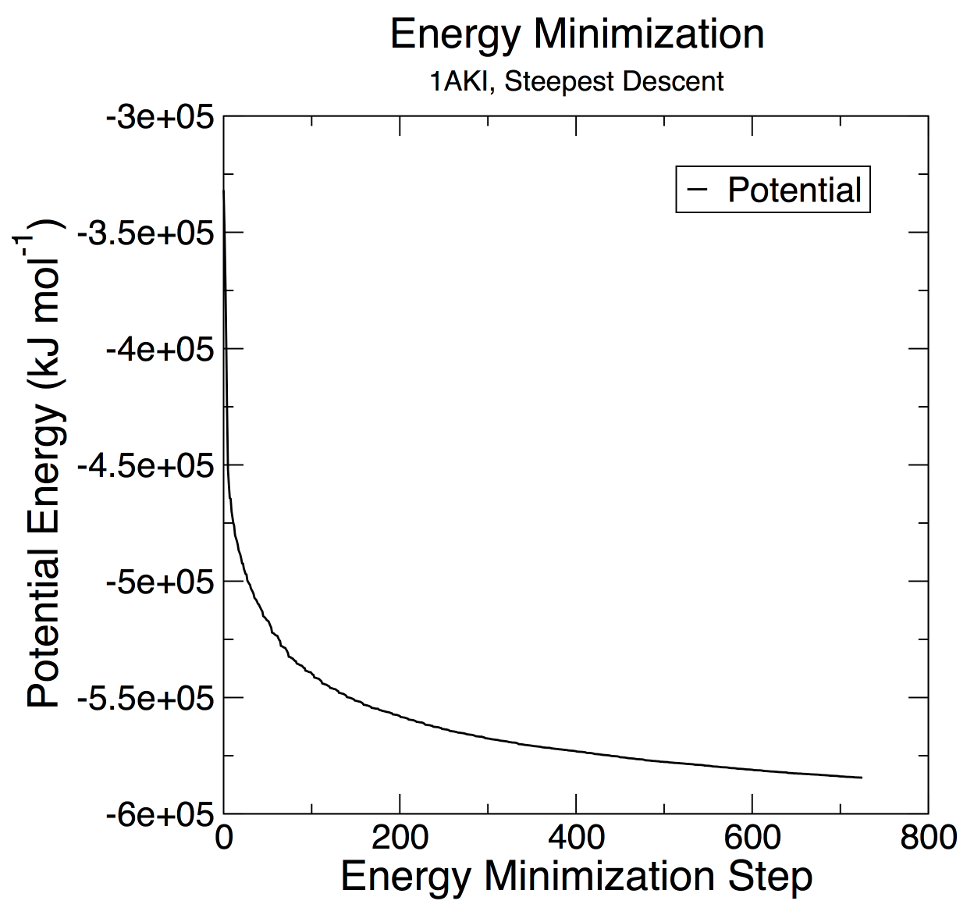
+NOTE:
+ The "Potential Energy" Epot should be negative, and (for a simple protein
+ in water) on the order of 105-106, depending on the system size and number
+ of water molecules.
+ The "Maximum force" Fmax should be no greater than the target for which was
+ set in minim.mdp - "emtol = 1000.0" in this case, no greater than 1000 kJ mol-1 nm-1
+
+This generates four files:
+
+```bash
+ls -l em.log em.trr em.edr em.gro
+```
+
+sample output:
+
+```
+-rw-rw-r-- 1 [CCRusername] nogroup 72368 Nov 6 11:00 em.edr
+-rw-rw-r-- 1 [CCRusername] nogroup 1788130 Nov 6 11:00 em.gro
+-rw-rw-r-- 1 [CCRusername] nogroup 185004 Nov 6 11:00 em.log
+-rw-rw-r-- 1 [CCRusername] nogroup 476940 Nov 6 11:00 em.trr
+```
+
+Analyze the .edr file "em.edr"
+
+```bash
+gmx energy -f em.edr -o potential.xvg
+```
+
+sample truncated output:
+
+```
+ :-) GROMACS - gmx energy, 2025.3 (-:
+
+Executable: /usr/local/gromacs/bin/gmx
+Data prefix: /usr/local/gromacs
+Working dir: /projects/academic/[YourGroupName]/GROMACS
+Command line:
+ gmx energy -f em.edr -o potential.xvg
+
+Opened em.edr as single precision energy file
+
+Select the terms you want from the following list by
+selecting either (part of) the name or the number or a combination.
+End your selection with an empty line or a zero.
+-------------------------------------------------------------------
+ 1 Bond 2 U-B 3 Proper-Dih. 4 Improper-Dih.
+ 5 CMAP-Dih. 6 LJ-14 7 Coulomb-14 8 LJ-(SR)
+ 9 Coulomb-(SR) 10 Coul.-recip. 11 Potential 12 Pressure
+ 13 Vir-XX 14 Vir-XY 15 Vir-XZ 16 Vir-YX
+[...]
+ 29 Pres-ZY 30 Pres-ZZ 31 #Surf*SurfTen 32 T-rest
+
+```
+
+Type "11 0" then [Enter] to select Potential (11); zero (0) terminate input
+
+```
+11 0
+```
+
+sample output:
+
+> ```
+> Last energy frame read 357 time 451.000
+>
+> Statistics over 452 steps [ 0.0000 through 451.0000 ps ], 1 data sets
+> All statistics are over 358 points (frames)
+>
+> Energy Average Err.Est. RMSD Tot-Drift
+> -------------------------------------------------------------------------------
+> Potential -601933 10000 24177.7 -67043.2 (kJ/mol)
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l potential.xvg
+```
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 11033 Nov 6 11:21 potential.xvg
+> ```
+
+The data in potential.xvg can be plotted, in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) with
+[xmgrace](https://plasma-gate.weizmann.ac.il/Grace/doc/UsersGuide.html#s3)
+
+Briefly:
+
+Launch a sesson in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) and connect
+to the desktop session
+[Applications] {Terminal Emulator]
+within the terminal window
+
+Change to your GROMACS directory
+
+```bash
+cd /projects/academic/[YourGroupName]/GROMACS
+```
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+Run "xmgrace" with the "potential.xvg" file
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ xmgrace "potential.xvg" \
+ -pexec 'title "Potential Energy"; subtitle "1AKI, Minimization with CHARMM36"; legend off; yaxis label "Potential Energy (kJ/mol\S-1\N)"; xaxis label "EM Step (ps)"'
+```
+
+This will display a plot that should look like this:
+
+
+Which is a little different to the sample output in the tutorial
+
+
+Please exit the OnDemand session once you are done:
+[Applications] [Log Out] [Log Out]
+Then close the browser window
+
+
+...back on the "salloc" interactive terminal session at the "Apptainer> " prompt
+
+
+Equilibrate the solvent and ions around the protein:
+Phase 1 is conducted under an NVT ensemble (constant Number of particles,
+Volume, and Temperature.)
+
+Download the .mdp file for this example:
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "./input/nvt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/nvt.mdp"
+```
+
+```bash
+gmx grompp -f inputs/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
+```
+
+Sample output:
+
+> ```
+> :-) GROMACS - gmx grompp, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx grompp -f inputs/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
+>
+> Ignoring obsolete mdp entry 'title'
+> Ignoring obsolete mdp entry 'ns_type'
+> Setting the LD random seed to -537190402
+>
+> Generated 167799 of the 167910 non-bonded parameter combinations
+> Generating 1-4 interactions: fudge = 1
+>
+> Generated 117432 of the 167910 1-4 parameter combinations
+>
+> Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
+>
+> turning H bonds into constraints...
+>
+> Excluding 2 bonded neighbours molecule type 'SOL'
+>
+> turning H bonds into constraints...
+>
+> Excluding 3 bonded neighbours molecule type 'CL'
+>
+> turning H bonds into constraints...
+>
+> Setting gen_seed to -718772497
+>
+> Velocities were taken from a Maxwell distribution at 300 K
+> Analysing residue names:
+> There are: 129 Protein residues
+> There are: 12589 Water residues
+> There are: 8 Ion residues
+> Analysing Protein...
+> Number of degrees of freedom in T-Coupling group Protein is 4920.82
+> Number of degrees of freedom in T-Coupling group non-Protein is 75555.18
+>
+> The largest distance between excluded atoms is 0.440 nm between atom 1156 and 1405
+>
+> Determining Verlet buffer for a tolerance of 0.005 kJ/mol/ps at 300 K
+>
+> Calculated rlist for 1x1 atom pair-list as 1.035 nm, buffer size 0.035 nm
+>
+> Set rlist, assuming 4x4 atom pair-list, to 1.000 nm, buffer size 0.000 nm
+>
+> Note that mdrun will redetermine rlist based on the actual pair-list setup
+>
+> NOTE 1 [file inputs/nvt.mdp]:
+> Removing center of mass motion in the presence of position restraints
+> might cause artifacts. When you are using position restraints to
+> equilibrate a macro-molecule, the artifacts are usually negligible.
+>
+> Calculating fourier grid dimensions for X Y Z
+> Using a fourier grid of 48x48x48, spacing 0.154 0.154 0.154
+>
+> Estimate for the relative computational load of the PME mesh part: 0.27
+>
+> This run will generate roughly 95 Mb of data
+>
+> There was 1 NOTE
+> [...]
+> ```
+
+This generates one file, "nvt.tpr" and updates mdout.mdp
+
+```bash
+ls -l nvt.tpr mdout.mdp
+```
+
+```
+-rw-rw-r-- 1 tkewtest nogroup 11031 Nov 6 14:36 mdout.mdp
+-rw-rw-r-- 1 tkewtest nogroup 1798084 Nov 6 14:36 nvt.tpr
+```
+
+Run the NVT simulation
+
+```bash
+gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm nvt
+```
+
+sample output:
+
+> ```
+> :-) GROMACS - gmx mdrun, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx mdrun -ntmpi 2 -deffnm nvt
+>
+> Reading file nvt.tpr, VERSION 2025.3 (single precision)
+> Changing nstlist from 10 to 50, rlist from 1 to 1.109
+> Using 1 MPI thread
+> Using 40 OpenMP threads
+>
+> starting mdrun 'LYSOZYME in water'
+> 50000 steps, 100.0 ps.
+>
+> Writing final coordinates.
+>
+> Core t (s) Wall t (s) (%)
+> Time: 4497.593 112.444 3999.8
+> (ns/day) (hour/ns)
+> Performance: 76.840 0.312
+> ```
+
+This generates five files:
+
+```bash
+ls -l nvt.cpt nvt.gro nvt.edr nvt.trr nvt.log
+```
+
+Sample output:
+
+```
+-rw-rw-r-- 1 [CCRusername] nogroup 955616 Nov 6 17:53 nvt.cpt
+-rw-rw-r-- 1 [CCRusername] nogroup 60104 Nov 6 17:53 nvt.edr
+-rw-rw-r-- 1 [CCRusername] nogroup 2741770 Nov 6 17:53 nvt.gro
+-rw-rw-r-- 1 [CCRusername] nogroup 95213 Nov 6 17:53 nvt.log
+-rw-rw-r-- 1 [CCRusername] nogroup 96329760 Nov 6 17:53 nvt.trr
+```
+
+Analyze the temperature progression
+
+```bash
+gmx energy -f nvt.edr -o temperature.xvg
+```
+
+Sample abridged output:
+
+> ```
+> :-) GROMACS - gmx energy, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx energy -f nvt.edr -o temperature.xvg
+>
+> Opened nvt.edr as single precision energy file
+>
+> Select the terms you want from the following list by
+> selecting either (part of) the name or the number or a combination.
+> End your selection with an empty line or a zero.
+> -------------------------------------------------------------------
+> 1 Bond 2 U-B 3 Proper-Dih. 4 Improper-Dih.
+> 5 CMAP-Dih. 6 LJ-14 7 Coulomb-14 8 LJ-(SR)
+> 9 Coulomb-(SR) 10 Coul.-recip. 11 Position-Rest. 12 Potential
+> 13 Kinetic-En. 14 Total-Energy 15 Conserved-En. 16 Temperature
+> [...]
+> 37 #Surf*SurfTen 38 T-System 39 Lamb-System
+>
+> ```
+
+Type "16 0" then [Enter] to select the temperature of the system (16); zero (0) terminate input
+
+```
+16 0
+```
+
+sample output:
+
+> ```
+> Last energy frame read 100 time 100.000
+>
+> Statistics over 50001 steps [ 0.0000 through 100.0000 ps ], 1 data sets
+> All statistics are over 501 points
+>
+> Energy Average Err.Est. RMSD Tot-Drift
+> -------------------------------------------------------------------------------
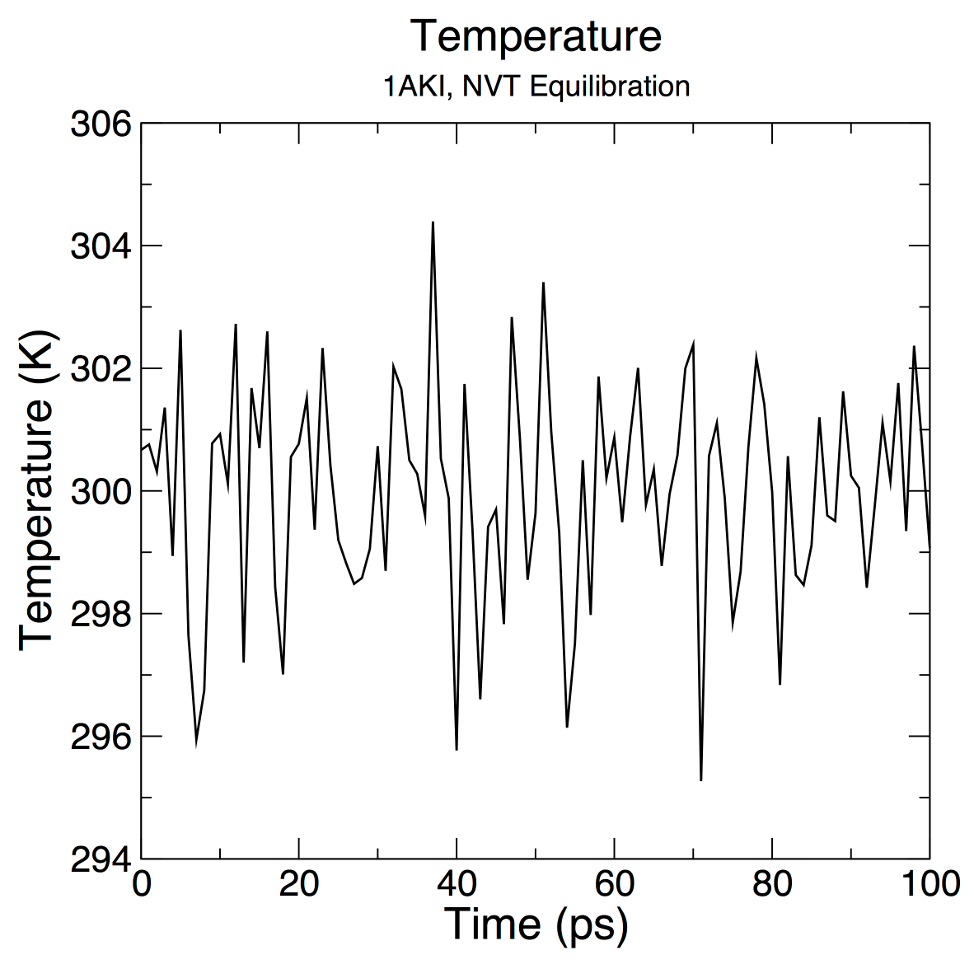
+> Temperature 299.812 0.12 3.15203 0.904259 (K)
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l temperature.xvg
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 3176 Nov 6 18:02 temperature.xvg
+> ```
+
+The data in temperature.xvg can be plotted, in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) similar
+to the above example for potential.xvg
+
+e.g. in an OnDemand terminal window
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+then
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ xmgrace "temperature.xvg" \
+ -pexec 'title "Temperature"; subtitle "1AKI, NVT Equilibration"; legend off; yaxis label "Temperature (K)"; s0 symbol 1'
+```
+
+My test plot looks like this:
+
+
+Which is a little different to the sample output in the tutorial
+
+
+Please exit the OnDemand session once you are done:
+[Applications] [Log Out] [Log Out]
+Then close the browser window
+
+
+...back on the "salloc" interactive terminal session at the "Apptainer> " prompt
+
+
+Equilibrate the solvent and ions around the protein:
+Phase 2 - Equilibration of pressure is conducted under an NPT ensemble where
+the Number of particles, Pressure, and Temperature are all constant
+
+Download the 500-ps NPT equilibration .mdp file "npt.mdp"
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "./input/npt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/npt.mdp"
+```
+
+```bash
+gmx grompp -f inputs/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
+```
+
+Sample output:
+
+> ```
+> :-) GROMACS - gmx grompp, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx grompp -f inputs/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
+>
+> Ignoring obsolete mdp entry 'title'
+> Ignoring obsolete mdp entry 'ns_type'
+> Setting the LD random seed to 1694439166
+>
+> Generated 167799 of the 167910 non-bonded parameter combinations
+> Generating 1-4 interactions: fudge = 1
+>
+> Generated 117432 of the 167910 1-4 parameter combinations
+>
+> Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
+>
+> turning H bonds into constraints...
+>
+> Excluding 2 bonded neighbours molecule type 'SOL'
+>
+> turning H bonds into constraints...
+>
+> Excluding 3 bonded neighbours molecule type 'CL'
+>
+> turning H bonds into constraints...
+>
+> Taking velocities from 'nvt.gro'
+>
+> NOTE 1 [file topol.top, line 18487]:
+> You are combining position restraints with Parrinello-Rahman pressure
+> coupling, which can lead to instabilities. If you really want to combine
+> position restraints with pressure coupling, we suggest to use C-rescale
+> pressure coupling instead.
+>
+> Analysing residue names:
+> There are: 129 Protein residues
+> There are: 12589 Water residues
+> There are: 8 Ion residues
+> Analysing Protein...
+> Number of degrees of freedom in T-Coupling group Protein is 4920.82
+> Number of degrees of freedom in T-Coupling group non-Protein is 75555.18
+>
+> The largest distance between excluded atoms is 0.446 nm between atom 1156 and 1405
+>
+> Determining Verlet buffer for a tolerance of 0.005 kJ/mol/ps at 300 K
+>
+> Calculated rlist for 1x1 atom pair-list as 1.035 nm, buffer size 0.035 nm
+>
+> Set rlist, assuming 4x4 atom pair-list, to 1.000 nm, buffer size 0.000 nm
+>
+> Note that mdrun will redetermine rlist based on the actual pair-list setup
+>
+> rest: 3.689 3.718 3.705
+> rest: 3.689 3.718 3.705
+>
+> NOTE 2 [file inputs/npt.mdp]:
+> Removing center of mass motion in the presence of position restraints
+> might cause artifacts. When you are using position restraints to
+> equilibrate a macro-molecule, the artifacts are usually negligible.
+>
+>
+> Reading Coordinates, Velocities and Box size from old trajectory
+>
+> Will read whole trajectory
+> Last frame -1 time 100.000
+>
+> Using frame at t = 100 ps
+>
+> Starting time for run is 0 ps
+> Calculating fourier grid dimensions for X Y Z
+> Using a fourier grid of 48x48x48, spacing 0.154 0.154 0.154
+>
+> Estimate for the relative computational load of the PME mesh part: 0.27
+>
+> This run will generate roughly 95 Mb of data
+>
+> There were 2 NOTEs
+> [...]
+> ```
+
+This generates one file, "npt.tpr" and updates mdout.mdp
+
+```bash
+ls -l npt.tpr mdout.mdp
+```
+
+Sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 11071 Nov 6 18:15 mdout.mdp
+> -rw-rw-r-- 1 [CCRusername] nogroup 1798108 Nov 6 18:15 npt.tpr
+> ```
+
+Run the NPT simulation
+
+This takes a couple of minutes to run on a node with 40 cores allocated:
+
+
+```bash
+gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm npt
+```
+
+sample output:
+
+> ```
+> :-) GROMACS - gmx mdrun, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx mdrun -ntmpi 2 -deffnm npt
+>
+> Reading file npt.tpr, VERSION 2025.3 (single precision)
+> Changing nstlist from 10 to 50, rlist from 1 to 1.109
+> Using 1 MPI thread
+> Using 40 OpenMP threads
+>
+> starting mdrun 'LYSOZYME in water'
+> 50000 steps, 100.0 ps.
+>
+> Writing final coordinates.
+>
+> Core t (s) Wall t (s) (%)
+> Time: 4621.352 115.544 3999.7
+> (ns/day) (hour/ns)
+> Performance: 74.778 0.321
+> [...]
+> ```
+
+This generates five files:
+
+```bash
+ls -l npt.cpt npt.gro npt.edr npt.trr npt.log
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 955600 Nov 6 18:17 npt.cpt
+> -rw-rw-r-- 1 [CCRusername] nogroup 307328 Nov 6 18:17 npt.edr
+> -rw-rw-r-- 1 [CCRusername] nogroup 2741563 Nov 6 18:17 npt.gro
+> -rw-rw-r-- 1 [CCRusername] nogroup 326363 Nov 6 18:17 npt.log
+> -rw-rw-r-- 1 [CCRusername] nogroup 477797688 Nov 6 18:17 npt.trr
+> ```
+
+
+Analyze the pressure progression
+
+```bash
+gmx energy -f npt.edr -o pressure.xvg
+```
+
+Sample abridged output:
+
+> ```
+> :-) GROMACS - gmx energy, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx energy -f npt.edr -o pressure.xvg
+>
+> Opened npt.edr as single precision energy file
+>
+> Select the terms you want from the following list by
+> selecting either (part of) the name or the number or a combination.
+> End your selection with an empty line or a zero.
+> -------------------------------------------------------------------
+> 1 Bond 2 U-B 3 Proper-Dih. 4 Improper-Dih.
+> 5 CMAP-Dih. 6 LJ-14 7 Coulomb-14 8 LJ-(SR)
+> 9 Coulomb-(SR) 10 Coul.-recip. 11 Position-Rest. 12 Potential
+> 13 Kinetic-En. 14 Total-Energy 15 Conserved-En. 16 Temperature
+> 17 Pressure 18 Constr.-rmsd 19 Box-X 20 Box-Y
+> [...]
+> 45 T-System 46 Lamb-System
+>
+> ```
+
+
+Type "17 0" then [Enter] to select the pressure of the system (17); zero (0) terminate input
+
+```
+17 0
+```
+
+sample output:
+
+> ```
+> Last energy frame read 100 time 100.000
+>
+> Statistics over 50001 steps [ 0.0000 through 100.0000 ps ], 1 data sets
+> All statistics are over 501 points
+>
+> Energy Average Err.Est. RMSD Tot-Drift
+> -------------------------------------------------------------------------------
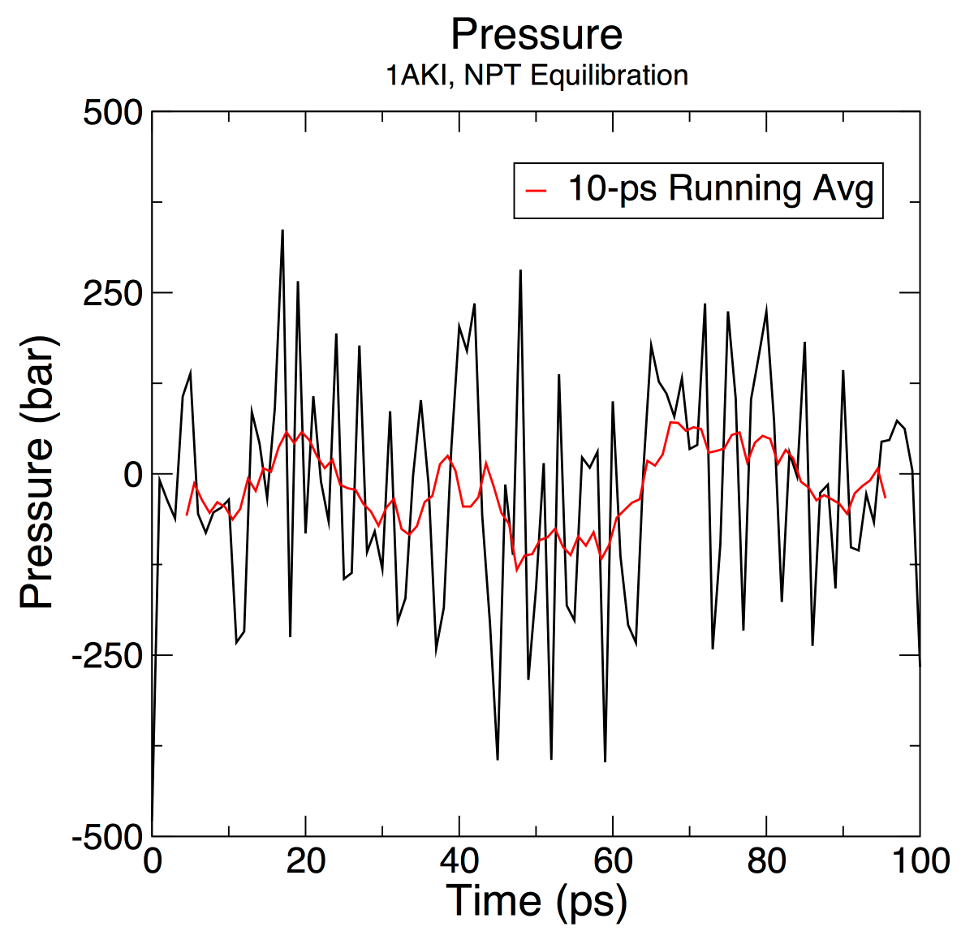
+> Pressure -1.63286 3.7 176.657 19.8596 (bar)
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l pressure.xvg
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 3175 Nov 7 14:10 pressure.xvg
+> ```
+
+The data in pressure.xvg can be plotted, in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) with
+"xmgrace"
+
+e.g. in an OnDemand terminal window
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+then
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ xmgrace "pressure.xvg" \
+ -pexec 'title "Pressure"; subtitle "1AKI, NPT Equilibration"; legend off; yaxis label "Pressure (bar)"; s0 line type 0; s0 symbol 1'
+```
+
+Add a 10th degree regression line with:
+
+[Data] [Transformations] [Regresssion...]
+Type of Fit: [10th Degree]
+[Accept]
+close the "Grace: Console" window and the "Regression" window
+
+
+My test plot looks like this:
+
+
+Which is a little different to the sample output in the tutorial
+
+
+Please exit the OnDemand session once you are done:
+[Applications] [Log Out] [Log Out]
+Then close the browser window
+
+
+...back on the "salloc" interactive terminal session at the "Apptainer> " prompt
+
+
+Examine the density using energy
+
+```bash
+gmx energy -f npt.edr -o density.xvg
+```
+
+Sample abridged output:
+
+> ```
+> :-) GROMACS - gmx energy, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx energy -f npt.edr -o density.xvg
+>
+> Opened npt.edr as single precision energy file
+>
+> Select the terms you want from the following list by
+> selecting either (part of) the name or the number or a combination.
+> End your selection with an empty line or a zero.
+> -------------------------------------------------------------------
+> 1 Bond 2 U-B 3 Proper-Dih. 4 Improper-Dih.
+> 5 CMAP-Dih. 6 LJ-14 7 Coulomb-14 8 LJ-(SR)
+> 9 Coulomb-(SR) 10 Coul.-recip. 11 Position-Rest. 12 Potential
+> 13 Kinetic-En. 14 Total-Energy 15 Conserved-En. 16 Temperature
+> 17 Pressure 18 Constr.-rmsd 19 Box-X 20 Box-Y
+> 21 Box-Z 22 Volume 23 Density 24 pV
+> [...]
+> 45 T-System 46 Lamb-System
+>
+> ```
+
+
+Type "23 0" then [Enter] to select the density of the system (23); zero (0) terminate input
+
+```
+23 0
+```
+
+sample output:
+
+> ```
+> Last energy frame read 500 time 500.000
+>
+> Statistics over 250001 steps [ 0.0000 through 500.0000 ps ], 1 data sets
+> All statistics are over 2501 points
+>
+> Energy Average Err.Est. RMSD Tot-Drift
+> -------------------------------------------------------------------------------
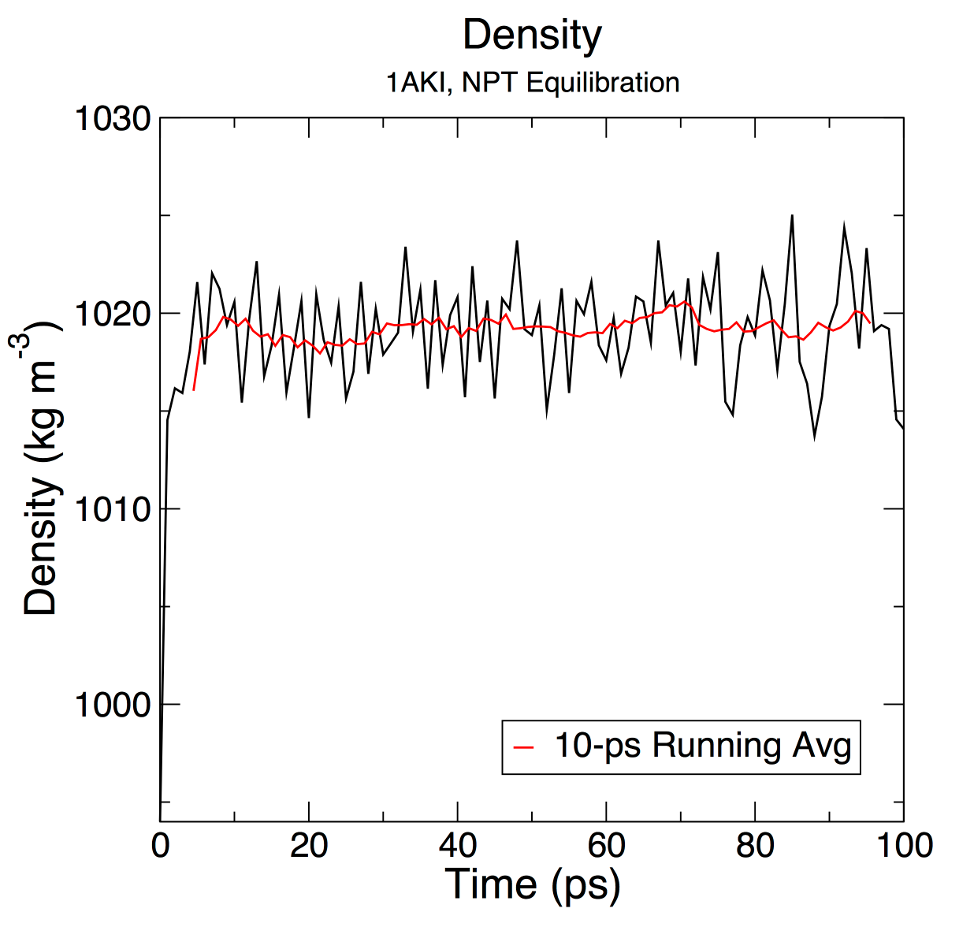
+> Density 996.782 0.31 2.27647 2.22393 (kg/m^3)
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l density.xvg
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 13247 Nov 11 09:57 density.xvg
+> ```
+
+The data in density.xvg can be plotted, in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) with
+"xmgrace"
+
+e.g. in an OnDemand terminal window
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+then
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ xmgrace "density.xvg" \
+ -pexec 'title "Density"; subtitle "1AKI, NPT Equilibration"; legend off; yaxis label "Density (kg m\S-3\N)";; s0 line type 0; s0 symbol 1'
+```
+
+Add a 10th degree regression line with:
+
+[Data] [Transformations] [Regresssion...]
+Type of Fit: [10th Degree]
+[Accept]
+close the "Grace: Console" window and the "Regression" window
+
+My test plot looks like this:
+
+
+Which is a little different to the sample output in the tutorial
+
+
+Please exit the OnDemand session once you are done:
+[Applications] [Log Out] [Log Out]
+Then close the browser window
+
+
+...back on the "salloc" interactive terminal session at the "Apptainer> " prompt
+
+
+Now the system is equilibrated, release the position restraints and run
+production MD for data collection
+
+Download the 10-ns MD simulation file (md.mdp) file from
+http://www.mdtutorials.com/
+
+Note: The "curl" command is commented out, because the file is already
+downloaded from the github repo
+
+```bash
+#curl -L -o "./input/md.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/md.mdp"
+```
+
+Generate the .tpr file for this simulation:
+
+```bash
+gmx grompp -f inputs/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_10.tpr
+```
+
+Sample output:
+
+> ```
+> :-) GROMACS - gmx grompp, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx grompp -f inputs/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_10.tpr
+>
+> Ignoring obsolete mdp entry 'title'
+> Ignoring obsolete mdp entry 'ns_type'
+> Setting the LD random seed to -593003529
+>
+> Generated 167799 of the 167910 non-bonded parameter combinations
+> Generating 1-4 interactions: fudge = 1
+>
+> Generated 117432 of the 167910 1-4 parameter combinations
+>
+> Excluding 3 bonded neighbours molecule type 'Protein_chain_A'
+>
+> turning H bonds into constraints...
+>
+> Excluding 2 bonded neighbours molecule type 'SOL'
+>
+> turning H bonds into constraints...
+>
+> Excluding 3 bonded neighbours molecule type 'CL'
+>
+> turning H bonds into constraints...
+>
+> Taking velocities from 'npt.gro'
+> Analysing residue names:
+> There are: 129 Protein residues
+> There are: 12589 Water residues
+> There are: 8 Ion residues
+> Analysing Protein...
+> Number of degrees of freedom in T-Coupling group Protein is 4920.82
+> Number of degrees of freedom in T-Coupling group non-Protein is 75555.18
+>
+> The largest distance between excluded atoms is 0.448 nm between atom 1156 and 1405
+>
+> Determining Verlet buffer for a tolerance of 0.005 kJ/mol/ps at 300 K
+>
+> Calculated rlist for 1x1 atom pair-list as 1.035 nm, buffer size 0.035 nm
+>
+> Set rlist, assuming 4x4 atom pair-list, to 1.000 nm, buffer size 0.000 nm
+>
+> Note that mdrun will redetermine rlist based on the actual pair-list setup
+>
+> Reading Coordinates, Velocities and Box size from old trajectory
+>
+> Will read whole trajectory
+> Last frame -1 time 100.000
+>
+> Using frame at t = 100 ps
+>
+> Starting time for run is 0 ps
+> Calculating fourier grid dimensions for X Y Z
+> Using a fourier grid of 48x48x48, spacing 0.152 0.152 0.152
+>
+> Estimate for the relative computational load of the PME mesh part: 0.27
+>
+> This run will generate roughly 23 Mb of data
+> [...]
+> ```
+
+
+This generates one file, "md_0_10.tpr" and updates mdout.mdp
+
+```bash
+ls -l md_0_10.tpr mdout.mdp
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 1690984 Nov 11 14:42 md_0_10.tpr
+> -rw-rw-r-- 1 [CCRusername] nogroup 10656 Nov 11 14:42 mdout.mdp
+> ```
+
+
+Run the 10-ns MD simulation:
+This takes about 20 minutes to run on a node with 40 cores allocated:
+
+```bash
+gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm md_0_10
+```
+
+Sample output:
+
+> ```
+> :-) GROMACS - gmx mdrun, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx mdrun -ntmpi 2 -deffnm md_0_10
+>
+> Reading file md_0_10.tpr, VERSION 2025.3 (single precision)
+> Changing nstlist from 10 to 50, rlist from 1 to 1.111
+> Using 1 MPI thread
+> Using 40 OpenMP threads
+>
+> starting mdrun 'LYSOZYME in water'
+> 500000 steps, 1000.0 ps.
+>
+> Writing final coordinates.
+>
+> Core t (s) Wall t (s) (%)
+> Core t (s) Wall t (s) (%)
+> Time: 46079.940 1152.002 4000.0
+> (ns/day) (hour/ns)
+> Performance: 75.000 0.320
+> [...]
+> ```
+
+This generates six files:
+
+```bash
+ls -l md_0_10.log md_0_10.xtc md_0_10.edr md_0_10.gro md_0_10_prev.cpt md_0_10.cpt
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 955976 Nov 11 15:40 md_0_10.cpt
+> -rw-rw-r-- 1 [CCRusername] nogroup 71140 Nov 11 15:40 md_0_10.edr
+> -rw-rw-r-- 1 [CCRusername] nogroup 2741770 Nov 11 15:40 md_0_10.gro
+> -rw-rw-r-- 1 [CCRusername] nogroup 92686 Nov 11 15:40 md_0_10.log
+> -rw-rw-r-- 1 [CCRusername] nogroup 955976 Nov 11 15:40 md_0_10_prev.cpt
+> -rw-rw-r-- 1 [CCRusername] nogroup 14426728 Nov 11 15:40 md_0_10.xtc
+> ```
+
+
+Correcting for Periodicity Effects
+Reimage the trajectory
+
+
+```bash
+gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_noPBC.xtc -pbc mol -center
+```
+
+Sample abridged output:
+
+> ```
+> :-) GROMACS - gmx trjconv, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_noPBC.xtc -pbc mol -center
+>
+> Note that major changes are planned in future for trjconv, to improve usability and utility.
+> Will write xtc: Compressed trajectory (portable xdr format): xtc
+> Reading file md_0_10.tpr, VERSION 2025.3 (single precision)
+> Reading file md_0_10.tpr, VERSION 2025.3 (single precision)
+> Select group for centering
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> [...]
+> Group 15 ( Ion) has 8 elements
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+Type "1" then [Enter] to select "Protein" at the "Select a group:" prompt
+
+```
+1
+```
+
+Sample abridged output:
+
+> ```
+> Selected 1: 'Protein'
+> Select group for output
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> [...]
+> Group 15 ( Ion) has 8 elements
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+Type "0" then [Enter] to select "System" for the output at the
+"Select a group:" prompt
+
+
+```
+0
+```
+
+Sample output:
+
+> ```
+> Selected 0: 'System'
+> Reading frame 0 time 0.000
+> Precision of md_0_10.xtc is 0.001 (nm)
+> Using output precision of 0.001 (nm)
+> Last frame 100 time 1000.000 -> frame 99 time 990.000
+> -> frame 100 time 1000.000
+> Last written: frame 100 time 1000.000
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l md_0_10_noPBC.xtc
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 14426744 Nov 11 16:16 md_0_10_noPBC.xtc
+> ```
+
+
+Root-Mean-Square Deviation
+
+```bash
+gmx rms -s md_0_10.tpr -f md_0_10_noPBC.xtc -o rmsd.xvg -tu ns
+```
+
+
+Sample abriged output:
+
+> ```
+> :-) GROMACS - gmx rms, 2025.3 (-:
+>
+> Executable: /usr/local/gromacs/bin/gmx
+> Data prefix: /usr/local/gromacs
+> Working dir: /projects/academic/[YourGroupName]/GROMACS
+> Command line:
+> gmx rms -s md_0_10.tpr -f md_0_10_noPBC.xtc -o rmsd.xvg -tu ns
+>
+> Reading file md_0_10.tpr, VERSION 2025.3 (single precision)
+> Reading file md_0_10.tpr, VERSION 2025.3 (single precision)
+> Select group for least squares fit
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> Group 3 ( C-alpha) has 129 elements
+> Group 4 ( Backbone) has 387 elements
+> Group 5 ( MainChain) has 515 elements
+> [...]
+> Group 15 ( Ion) has 8 elements
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+
+Type "4" then [Enter] to select "Backbone" for the least-squares fit at the
+"Select a group:" prompt
+
+```
+4
+```
+
+Sample abriged output:
+
+> ```
+> Selected 4: 'Backbone'
+> Select group for RMSD calculation
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> Group 3 ( C-alpha) has 129 elements
+> Group 4 ( Backbone) has 387 elements
+> Group 5 ( MainChain) has 515 elements
+> [...]
+> Group 15 ( Ion) has 8 elements
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+Type "4" then [Enter] to select "Backbone" for the group for RMSD calculation
+"Select a group:" prompt
+
+```
+4
+```
+
+Sample output:
+
+> ```
+> Selected 4: 'Backbone'
+> Last frame 100 time 1.000
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l rmsd.xvg
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 3207 Nov 11 16:35 rmsd.xvg
+> ```
+
+The data in this .xvg file, and the one that follows, can be can be plotted,
+in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) with "xmgrace"
+
+
+Calculate RMSD relative to the crystal structure
+
+```bash
+gmx rms -s em.tpr -f md_0_10_noPBC.xtc -o rmsd_xtal.xvg -tu ns
+```
+
+Sample abridged output:
+
+> ```
+> [...]
+> Reading file em.tpr, VERSION 2025.3 (single precision)
+> Reading file em.tpr, VERSION 2025.3 (single precision)
+> Select group for least squares fit
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> Group 3 ( C-alpha) has 129 elements
+> Group 4 ( Backbone) has 387 elements
+> Group 5 ( MainChain) has 515 elements
+> [...]
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+Type "4" then [Enter] to select "Backbone" for the least-squares fit at the
+"Select a group:" prompt
+
+```
+4
+```
+
+Sample abriged output:
+
+> ```
+> Selected 4: 'Backbone'
+> Select group for RMSD calculation
+> Group 0 ( System) has 39735 elements
+> Group 1 ( Protein) has 1960 elements
+> Group 2 ( Protein-H) has 1001 elements
+> Group 3 ( C-alpha) has 129 elements
+> Group 4 ( Backbone) has 387 elements
+> Group 5 ( MainChain) has 515 elements
+> [...]
+> Group 16 ( Water_and_ions) has 37775 elements
+> Select a group:
+> ```
+
+Type "4" then [Enter] to select "Backbone" for the group for RMSD calculation
+"Select a group:" prompt
+
+```
+4
+```
+
+Sample output:
+
+> ```
+> Selected 4: 'Backbone'
+> Last frame 100 time 1.000
+> [...]
+> ```
+
+This generates one file
+
+```bash
+ls -l rmsd_xtal.xvg
+```
+
+sample output:
+
+> ```
+> -rw-rw-r-- 1 [CCRusername] nogroup 3197 Nov 11 16:52 rmsd_xtal.xvg
+> ```
+
+
+The data in this .xvg file, and the prior one (rmsd.xvg), can be can be
+plotted, in CCR's [OnDemand portal](https://ondemand.ccr.buffalo.edu) with "xmgrace"
+
+e.g. in an OnDemand terminal window
+
+Set the directory for container images e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+then
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ xmgrace "rmsd.xvg" "rmsd_xtal.xvg" \
+ -pexec 'title "RMSD"; subtitle "1AKI, Backbone"; legend on; s0 line type 0; s0 symbol 1; s1 line type 0; s1 symbol 2'
+```
+Add 10th degree regression lines with:
+
+[Data] [Transformations] [Regresssion...]
+click on "All sets"
+Type of Fit: [10th Degree]
+[Accept]
+close the "Grace: Console" window and the "Regression" window
+
+Label the two regressions:
+
+[Plot] [Set Apprearance]
+select "G0.S2[2][101]"
+Legend Sring: "Ref: Equilibrated"
+[Apply]
+select "G0.S3[2][101]"
+Legend Sring: "Ref: Crystal"
+[Accept]
+
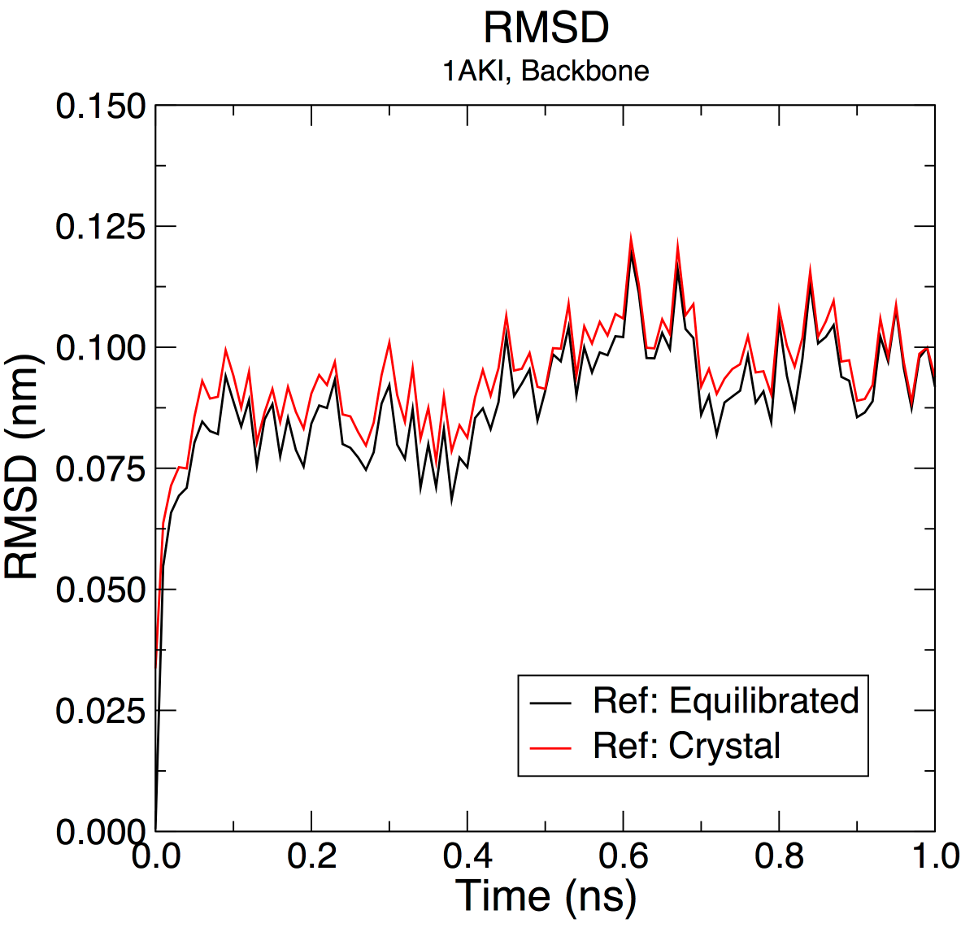
+My test plot looks like this:
+
+
+Which is a little different to the sample output in the tutorial
+
+
+Please exit the OnDemand session once you are done:
+[Applications] [Log Out] [Log Out]
+Then close the browser window
+
+
+...back on the "salloc" interactive terminal session at the "Apptainer> " prompt
+
+Exit the container
+
+```bash
+exit
+```
+
+sample output:
+
+> ```bash
+> CCRusername@cpn-c04-33:/projects/academic/[YourGroupName]/GROMACS$
+> ``
+
+...then exit the slurm interactive session
+
+```bash
+exit
+```
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/README.md b/containers/2_ApplicationSpecific/GROMACS/README.md
new file mode 100644
index 00000000..de4649ba
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/README.md
@@ -0,0 +1,120 @@
+# Examples for using nvidia's GROMACS container
+
+GROMACS is an open-source software suite for high-performance molecular dynamics and output analysis.
+
+The interactive and Slurm examples use nvidia's container.
+The container supports nvidia GPUs, but is limited to a single node.
+
+The interactive examples follow the [Lysozyme in Water GROMACS tutorial](http://www.mdtutorials.com/gmx/lysozyme/01_pdb2gmx.html)
+and the long Slurm example script does the same computations.
+
+The interacrive examples use "xmgrace" - instructions for buildind a grace
+container are below.
+This is NOT required to use nvidia's GROMACS container. The sample Slurm
+scrtips do NOT need this.
+
+
+## Building a container with grace/xmgrace
+
+1. Start an interactive job in the debug queue
+see the [CCR documantation on Slurm jobs](https://docs.ccr.buffalo.edu/en/latest/hpc/jobs) for more information
+
+e.g.
+
+```bash
+salloc --cluster=ub-hpc --partition=debug --qos=debug --mem=0 --exclusive --time=01:00:00
+```
+
+2. Change to your build directory
+e.g.
+
+```bash
+cd /projects/academic/[YourGroupName]/GROMACS
+```
+
+Set the target directory for all your container images
+e.g.
+
+```bash
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+```
+
+Make sure APPTAINER_TMPDIR and APPTAINER_CACHEDIR environment
+variables are set to sensible values
+
+```bash
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+export APPTAINER_CACHEDIR="${APPTAINER_CACHEDIR:-${SLURMTMPDIR}/apptainer}"
+mkdir -p "${APPTAINER_CACHEDIR}"
+```
+
+Download the .def file
+
+```bash
+#curl -L -o "grace.def" "https://raw.githubusercontent.com/tonykew/ccr-examples/refs/heads/GROMACS/containers/2_ApplicationSpecific/GROMACS/grace.def"
+curl -L -o "grace.def" "https://raw.githubusercontent.com/ubccr/ccr-examples/refs/heads/main/containers/2_ApplicationSpecific/GROMACS/grace.def"
+```
+
+Build the grace container image
+
+```bash
+apptainer build "${CONTAINER_DIR}/grace-$(arch).sif" "grace.def"
+```
+
+Verify that the build was successful
+
+```bash
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/grace-$(arch).sif" \
+ grace -version
+```
+
+Sample output:
+
+>
+> Grace-5.1.25
+>
+> GUI toolkit: @(#)Motif Version 2.3.8
+> Xbae version: 46004
+> T1lib: 1.3.1p3-grace
+> FFT: FFTW
+> NetCDF support: on
+> libpng: 1.6.43
+> libjpeg: 80
+> Built: Mon Apr 8 10:40:41 2024 on Linux #191-Ubuntu SMP Fri Feb 2 13:55:07 UTC 2024 5.4.0-173-generic x86_64
+> Compiler flags: gcc -g -O2 -fno-omit-frame-pointer -mno-omit-leaf-frame-pointer -ffile-prefix-map=/build/grace-mFwzxU/grace-5.1.25=. -fstack-protector-strong -fstack-clash-protection -Wformat -Werror=format-security -fcf-protection -fdebug-prefix-map=/build/grace-mFwzxU/grace-5.1.25=/usr/src/grace-1:5.1.25-14 -I.. -I. -I../T1lib/t1lib -Wdate-time -D_FORTIFY_SOURCE=3 -Wl,-Bsymbolic-functions -Wl,-z,relro -Wl,-z,now -lXmHTML -lXbae -lXm -lXpm -lXmu -lXt -lXext -lX11 -lSM -lICE ../cephes/libcephes.a -lnetcdf -lfftw3 ../T1lib/libt1.a -ljpeg -lpng -lz -ltirpc -lm
+>
+> Registered devices:
+> Dummy PostScript EPS MIF SVG PNM JPEG PNG Metafile
+>
+> (C) Copyright 1991-1995 Paul J Turner
+> (C) Copyright 1996-2015 Grace Development Team
+> All Rights Reserved
+
+
+## Example Scripts
+
+Provided in this repository are a couple of example GROMACS Slurm jobs.
+
+### Short Slurm example
+
+x86_64
+[slurm_nvidia_GROMACS_short_example.bash](./slurm_nvidia_GROMACS_short_example.bash)
+
+ARM64
+[slurm_ARM64_nvidia_GROMACS_short_example.bash](./slurm_ARM64_nvidia_GROMACS_short_example.bash)
+
+
+### Long Slurm example
+
+This example does all the computations from the [Lysozyme in Water GROMACS tutorial](http://www.mdtutorials.com/gmx/lysozyme/01_pdb2gmx.html)
+
+x86_64
+[slurm_nvidia_GROMACS_long_example.bash](./slurm_nvidia_GROMACS_long_example.bash)
+
+ARM64
+[slurm_ARM64_nvidia_GROMACS_long_example.bash](./slurm_ARM64_nvidia_GROMACS_long_example.bash)
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/charmm36-jul2022.ff.tgz b/containers/2_ApplicationSpecific/GROMACS/charmm36-jul2022.ff.tgz
new file mode 100644
index 00000000..8c0c3345
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/charmm36-jul2022.ff.tgz differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/grace.def b/containers/2_ApplicationSpecific/GROMACS/grace.def
new file mode 100644
index 00000000..3bd2f169
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/grace.def
@@ -0,0 +1,61 @@
+Bootstrap: docker
+From: ubuntu:24.04
+
+%post -c /bin/bash
+ # Set the timezone, if unset
+ test -h /etc/localtime || ln -fs /usr/share/zoneinfo/America/New_York /etc/localtime
+
+ cp /etc/apt/sources.list /etc/apt/sources.list~
+ sed -E -i 's/^# deb-src /deb-src /' /etc/apt/sources.list
+ apt-get -y update
+
+ # Install man & man pages - this section can be removed if not needed
+ # NOTE: Do this before installing anything else so their man pages are installed
+ sed -e '\|/usr/share/man|s|^#*|#|g' -i /etc/dpkg/dpkg.cfg.d/excludes
+ DEBIAN_FRONTEND=noninteractive apt-get -y install apt-utils groff dialog man-db manpages manpages-posix manpages-dev
+ rm -f /usr/bin/man
+ dpkg-divert --quiet --remove --rename /usr/bin/man
+
+ # O/S package updates:
+ DEBIAN_FRONTEND=noninteractive apt-get -y upgrade
+
+ DEBIAN_FRONTEND=noninteractive apt-get -y install \
+ tzdata \
+ locales \
+ unzip \
+ wget \
+ git \
+ gawk \
+ pax-utils \
+ python3 \
+ python3-sphinx \
+ python3-sphinx-rtd-theme \
+ python-is-python3 \
+ grace \
+ curl \
+ less \
+ jq \
+ nano \
+ vim \
+ apt-file
+
+ # NOTE: apt-file is generally not needed to run, but can be useful during development
+ apt-file update
+
+ # These steps are necessary to configure Perl and can cause issues with Python if omitted
+ sed -i -e 's/# en_US.UTF-8 UTF-8/en_US.UTF-8 UTF-8/' /etc/locale.gen
+ dpkg-reconfigure --frontend=noninteractive locales
+ update-locale LANG=en_US.UTF-8
+
+%environment
+ export LANG=en_US.UTF-8
+ # Change the nvidia cache dir from ~/.nv/ComputeCache
+ export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+ mkdir -p "${CUDA_CACHE_PATH}"
+
+%runscript
+ #!/bin/bash
+ export PS1="xmgrace$ "
+ # Exec passed command (required for Modal ENTRYPOINT compatibility)
+ exec "$@"
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/Untitled.png b/containers/2_ApplicationSpecific/GROMACS/images/Untitled.png
new file mode 100644
index 00000000..b3f11d10
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/Untitled.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/density.png b/containers/2_ApplicationSpecific/GROMACS/images/density.png
new file mode 100644
index 00000000..8f332785
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/density.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/potential.png b/containers/2_ApplicationSpecific/GROMACS/images/potential.png
new file mode 100644
index 00000000..a2edf24e
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/potential.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/pressure.png b/containers/2_ApplicationSpecific/GROMACS/images/pressure.png
new file mode 100644
index 00000000..65290abe
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/pressure.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/rmsd_xtal.png b/containers/2_ApplicationSpecific/GROMACS/images/rmsd_xtal.png
new file mode 100644
index 00000000..29111e80
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/rmsd_xtal.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/images/temperature.png b/containers/2_ApplicationSpecific/GROMACS/images/temperature.png
new file mode 100644
index 00000000..47227230
Binary files /dev/null and b/containers/2_ApplicationSpecific/GROMACS/images/temperature.png differ
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/1AKI.pdb b/containers/2_ApplicationSpecific/GROMACS/inputs/1AKI.pdb
new file mode 100644
index 00000000..4ffefabe
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/1AKI.pdb
@@ -0,0 +1,1437 @@
+HEADER HYDROLASE 19-MAY-97 1AKI
+TITLE THE STRUCTURE OF THE ORTHORHOMBIC FORM OF HEN EGG-WHITE LYSOZYME AT
+TITLE 2 1.5 ANGSTROMS RESOLUTION
+COMPND MOL_ID: 1;
+COMPND 2 MOLECULE: LYSOZYME;
+COMPND 3 CHAIN: A;
+COMPND 4 EC: 3.2.1.17
+SOURCE MOL_ID: 1;
+SOURCE 2 ORGANISM_SCIENTIFIC: GALLUS GALLUS;
+SOURCE 3 ORGANISM_COMMON: CHICKEN;
+SOURCE 4 ORGANISM_TAXID: 9031;
+SOURCE 5 CELL: EGG
+KEYWDS HYDROLASE, GLYCOSIDASE
+EXPDTA X-RAY DIFFRACTION
+AUTHOR D.CARTER,J.HE,J.R.RUBLE,B.WRIGHT
+REVDAT 4 20-NOV-24 1AKI 1 REMARK
+REVDAT 3 02-AUG-23 1AKI 1 REMARK
+REVDAT 2 24-FEB-09 1AKI 1 VERSN
+REVDAT 1 19-NOV-97 1AKI 0
+JRNL AUTH P.J.ARTYMIUK,C.C.F.BLAKE,D.W.RICE,K.S.WILSON
+JRNL TITL THE STRUCTURES OF THE MONOCLINIC AND ORTHORHOMBIC FORMS OF
+JRNL TITL 2 HEN EGG-WHITE LYSOZYME AT 6 ANGSTROMS RESOLUTION
+JRNL REF ACTA CRYSTALLOGR.,SECT.B V. 38 778 1982
+JRNL REFN ISSN 0108-7681
+REMARK 2
+REMARK 2 RESOLUTION. 1.50 ANGSTROMS.
+REMARK 3
+REMARK 3 REFINEMENT.
+REMARK 3 PROGRAM : GPRLSA
+REMARK 3 AUTHORS : FUREY
+REMARK 3
+REMARK 3 DATA USED IN REFINEMENT.
+REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 1.50
+REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : 10.00
+REMARK 3 DATA CUTOFF (SIGMA(F)) : 1.000
+REMARK 3 COMPLETENESS FOR RANGE (%) : 91.1
+REMARK 3 NUMBER OF REFLECTIONS : 16327
+REMARK 3
+REMARK 3 FIT TO DATA USED IN REFINEMENT.
+REMARK 3 CROSS-VALIDATION METHOD : NULL
+REMARK 3 FREE R VALUE TEST SET SELECTION : NULL
+REMARK 3 R VALUE (WORKING + TEST SET) : NULL
+REMARK 3 R VALUE (WORKING SET) : 0.212
+REMARK 3 FREE R VALUE : NULL
+REMARK 3 FREE R VALUE TEST SET SIZE (%) : NULL
+REMARK 3 FREE R VALUE TEST SET COUNT : NULL
+REMARK 3
+REMARK 3 FIT/AGREEMENT OF MODEL WITH ALL DATA.
+REMARK 3 R VALUE (WORKING + TEST SET, NO CUTOFF) : NULL
+REMARK 3 R VALUE (WORKING SET, NO CUTOFF) : NULL
+REMARK 3 FREE R VALUE (NO CUTOFF) : NULL
+REMARK 3 FREE R VALUE TEST SET SIZE (%, NO CUTOFF) : NULL
+REMARK 3 FREE R VALUE TEST SET COUNT (NO CUTOFF) : NULL
+REMARK 3 TOTAL NUMBER OF REFLECTIONS (NO CUTOFF) : NULL
+REMARK 3
+REMARK 3 NUMBER OF NON-HYDROGEN ATOMS USED IN REFINEMENT.
+REMARK 3 PROTEIN ATOMS : 1001
+REMARK 3 NUCLEIC ACID ATOMS : 0
+REMARK 3 HETEROGEN ATOMS : 0
+REMARK 3 SOLVENT ATOMS : 78
+REMARK 3
+REMARK 3 B VALUES.
+REMARK 3 FROM WILSON PLOT (A**2) : NULL
+REMARK 3 MEAN B VALUE (OVERALL, A**2) : NULL
+REMARK 3 OVERALL ANISOTROPIC B VALUE.
+REMARK 3 B11 (A**2) : NULL
+REMARK 3 B22 (A**2) : NULL
+REMARK 3 B33 (A**2) : NULL
+REMARK 3 B12 (A**2) : NULL
+REMARK 3 B13 (A**2) : NULL
+REMARK 3 B23 (A**2) : NULL
+REMARK 3
+REMARK 3 ESTIMATED COORDINATE ERROR.
+REMARK 3 ESD FROM LUZZATI PLOT (A) : NULL
+REMARK 3 ESD FROM SIGMAA (A) : NULL
+REMARK 3 LOW RESOLUTION CUTOFF (A) : 10.0
+REMARK 3
+REMARK 3 RMS DEVIATIONS FROM IDEAL VALUES.
+REMARK 3 DISTANCE RESTRAINTS. RMS SIGMA
+REMARK 3 BOND LENGTH (A) : 0.009 ; 0.010
+REMARK 3 ANGLE DISTANCE (A) : 0.003 ; 0.025
+REMARK 3 INTRAPLANAR 1-4 DISTANCE (A) : 0.024 ; 0.020
+REMARK 3 H-BOND OR METAL COORDINATION (A) : NULL ; NULL
+REMARK 3
+REMARK 3 PLANE RESTRAINT (A) : 0.033 ; 0.030
+REMARK 3 CHIRAL-CENTER RESTRAINT (A**3) : 0.212 ; 0.200
+REMARK 3
+REMARK 3 NON-BONDED CONTACT RESTRAINTS.
+REMARK 3 SINGLE TORSION (A) : 0.183 ; 0.300
+REMARK 3 MULTIPLE TORSION (A) : 0.159 ; 0.300
+REMARK 3 H-BOND (X...Y) (A) : 0.299 ; 0.300
+REMARK 3 H-BOND (X-H...Y) (A) : NULL ; NULL
+REMARK 3
+REMARK 3 CONFORMATIONAL TORSION ANGLE RESTRAINTS.
+REMARK 3 SPECIFIED (DEGREES) : NULL ; NULL
+REMARK 3 PLANAR (DEGREES) : 7.900 ; 5.000
+REMARK 3 STAGGERED (DEGREES) : 17.800; 15.000
+REMARK 3 TRANSVERSE (DEGREES) : 18.900; 15.000
+REMARK 3
+REMARK 3 ISOTROPIC THERMAL FACTOR RESTRAINTS. RMS SIGMA
+REMARK 3 MAIN-CHAIN BOND (A**2) : 2.500 ; 3.000
+REMARK 3 MAIN-CHAIN ANGLE (A**2) : 2.900 ; 4.000
+REMARK 3 SIDE-CHAIN BOND (A**2) : 3.200 ; 4.000
+REMARK 3 SIDE-CHAIN ANGLE (A**2) : 3.600 ; 3.000
+REMARK 3
+REMARK 3 OTHER REFINEMENT REMARKS: NULL
+REMARK 4
+REMARK 4 1AKI COMPLIES WITH FORMAT V. 3.30, 13-JUL-11
+REMARK 100
+REMARK 100 THIS ENTRY HAS BEEN PROCESSED BY BNL.
+REMARK 100 THE DEPOSITION ID IS D_1000170929.
+REMARK 200
+REMARK 200 EXPERIMENTAL DETAILS
+REMARK 200 EXPERIMENT TYPE : X-RAY DIFFRACTION
+REMARK 200 DATE OF DATA COLLECTION : NOV-95
+REMARK 200 TEMPERATURE (KELVIN) : 298
+REMARK 200 PH : 4.48
+REMARK 200 NUMBER OF CRYSTALS USED : 1
+REMARK 200
+REMARK 200 SYNCHROTRON (Y/N) : N
+REMARK 200 RADIATION SOURCE : ROTATING ANODE
+REMARK 200 BEAMLINE : NULL
+REMARK 200 X-RAY GENERATOR MODEL : RIGAKU RUH2R
+REMARK 200 MONOCHROMATIC OR LAUE (M/L) : M
+REMARK 200 WAVELENGTH OR RANGE (A) : 1.5418
+REMARK 200 MONOCHROMATOR : GRAPHITE(002)
+REMARK 200 OPTICS : NULL
+REMARK 200
+REMARK 200 DETECTOR TYPE : IMAGE PLATE
+REMARK 200 DETECTOR MANUFACTURER : RIGAKU RAXIS IIC
+REMARK 200 INTENSITY-INTEGRATION SOFTWARE : BIOTEX, RIGAKU
+REMARK 200 DATA SCALING SOFTWARE : BIOTEX
+REMARK 200
+REMARK 200 NUMBER OF UNIQUE REFLECTIONS : 20571
+REMARK 200 RESOLUTION RANGE HIGH (A) : 1.500
+REMARK 200 RESOLUTION RANGE LOW (A) : 15.000
+REMARK 200 REJECTION CRITERIA (SIGMA(I)) : 1.000
+REMARK 200
+REMARK 200 OVERALL.
+REMARK 200 COMPLETENESS FOR RANGE (%) : 91.1
+REMARK 200 DATA REDUNDANCY : 3.100
+REMARK 200 R MERGE (I) : 0.04400
+REMARK 200 R SYM (I) : NULL
+REMARK 200 FOR THE DATA SET : 11.7000
+REMARK 200
+REMARK 200 IN THE HIGHEST RESOLUTION SHELL.
+REMARK 200 HIGHEST RESOLUTION SHELL, RANGE HIGH (A) : NULL
+REMARK 200 HIGHEST RESOLUTION SHELL, RANGE LOW (A) : NULL
+REMARK 200 COMPLETENESS FOR SHELL (%) : NULL
+REMARK 200 DATA REDUNDANCY IN SHELL : NULL
+REMARK 200 R MERGE FOR SHELL (I) : NULL
+REMARK 200 R SYM FOR SHELL (I) : NULL
+REMARK 200 FOR SHELL : NULL
+REMARK 200
+REMARK 200 DIFFRACTION PROTOCOL: NULL
+REMARK 200 METHOD USED TO DETERMINE THE STRUCTURE: MOLECULAR REPLACEMENT
+REMARK 200 SOFTWARE USED: X-PLOR
+REMARK 200 STARTING MODEL: PDB ENTRY 2LZH
+REMARK 200
+REMARK 200 REMARK: NULL
+REMARK 280
+REMARK 280 CRYSTAL
+REMARK 280 SOLVENT CONTENT, VS (%): 42.84
+REMARK 280 MATTHEWS COEFFICIENT, VM (ANGSTROMS**3/DA): 2.15
+REMARK 280
+REMARK 280 CRYSTALLIZATION CONDITIONS: PH 4.48
+REMARK 290
+REMARK 290 CRYSTALLOGRAPHIC SYMMETRY
+REMARK 290 SYMMETRY OPERATORS FOR SPACE GROUP: P 21 21 21
+REMARK 290
+REMARK 290 SYMOP SYMMETRY
+REMARK 290 NNNMMM OPERATOR
+REMARK 290 1555 X,Y,Z
+REMARK 290 2555 -X+1/2,-Y,Z+1/2
+REMARK 290 3555 -X,Y+1/2,-Z+1/2
+REMARK 290 4555 X+1/2,-Y+1/2,-Z
+REMARK 290
+REMARK 290 WHERE NNN -> OPERATOR NUMBER
+REMARK 290 MMM -> TRANSLATION VECTOR
+REMARK 290
+REMARK 290 CRYSTALLOGRAPHIC SYMMETRY TRANSFORMATIONS
+REMARK 290 THE FOLLOWING TRANSFORMATIONS OPERATE ON THE ATOM/HETATM
+REMARK 290 RECORDS IN THIS ENTRY TO PRODUCE CRYSTALLOGRAPHICALLY
+REMARK 290 RELATED MOLECULES.
+REMARK 290 SMTRY1 1 1.000000 0.000000 0.000000 0.00000
+REMARK 290 SMTRY2 1 0.000000 1.000000 0.000000 0.00000
+REMARK 290 SMTRY3 1 0.000000 0.000000 1.000000 0.00000
+REMARK 290 SMTRY1 2 -1.000000 0.000000 0.000000 29.53100
+REMARK 290 SMTRY2 2 0.000000 -1.000000 0.000000 0.00000
+REMARK 290 SMTRY3 2 0.000000 0.000000 1.000000 15.25850
+REMARK 290 SMTRY1 3 -1.000000 0.000000 0.000000 0.00000
+REMARK 290 SMTRY2 3 0.000000 1.000000 0.000000 34.22550
+REMARK 290 SMTRY3 3 0.000000 0.000000 -1.000000 15.25850
+REMARK 290 SMTRY1 4 1.000000 0.000000 0.000000 29.53100
+REMARK 290 SMTRY2 4 0.000000 -1.000000 0.000000 34.22550
+REMARK 290 SMTRY3 4 0.000000 0.000000 -1.000000 0.00000
+REMARK 290
+REMARK 290 REMARK: NULL
+REMARK 300
+REMARK 300 BIOMOLECULE: 1
+REMARK 300 SEE REMARK 350 FOR THE AUTHOR PROVIDED AND/OR PROGRAM
+REMARK 300 GENERATED ASSEMBLY INFORMATION FOR THE STRUCTURE IN
+REMARK 300 THIS ENTRY. THE REMARK MAY ALSO PROVIDE INFORMATION ON
+REMARK 300 BURIED SURFACE AREA.
+REMARK 350
+REMARK 350 COORDINATES FOR A COMPLETE MULTIMER REPRESENTING THE KNOWN
+REMARK 350 BIOLOGICALLY SIGNIFICANT OLIGOMERIZATION STATE OF THE
+REMARK 350 MOLECULE CAN BE GENERATED BY APPLYING BIOMT TRANSFORMATIONS
+REMARK 350 GIVEN BELOW. BOTH NON-CRYSTALLOGRAPHIC AND
+REMARK 350 CRYSTALLOGRAPHIC OPERATIONS ARE GIVEN.
+REMARK 350
+REMARK 350 BIOMOLECULE: 1
+REMARK 350 AUTHOR DETERMINED BIOLOGICAL UNIT: MONOMERIC
+REMARK 350 APPLY THE FOLLOWING TO CHAINS: A
+REMARK 350 BIOMT1 1 1.000000 0.000000 0.000000 0.00000
+REMARK 350 BIOMT2 1 0.000000 1.000000 0.000000 0.00000
+REMARK 350 BIOMT3 1 0.000000 0.000000 1.000000 0.00000
+REMARK 500
+REMARK 500 GEOMETRY AND STEREOCHEMISTRY
+REMARK 500 SUBTOPIC: CLOSE CONTACTS IN SAME ASYMMETRIC UNIT
+REMARK 500
+REMARK 500 THE FOLLOWING ATOMS ARE IN CLOSE CONTACT.
+REMARK 500
+REMARK 500 ATM1 RES C SSEQI ATM2 RES C SSEQI DISTANCE
+REMARK 500 NH2 ARG A 45 NH2 ARG A 68 2.16
+REMARK 500
+REMARK 500 REMARK: NULL
+REMARK 500
+REMARK 500 GEOMETRY AND STEREOCHEMISTRY
+REMARK 500 SUBTOPIC: CLOSE CONTACTS
+REMARK 500
+REMARK 500 THE FOLLOWING ATOMS THAT ARE RELATED BY CRYSTALLOGRAPHIC
+REMARK 500 SYMMETRY ARE IN CLOSE CONTACT. AN ATOM LOCATED WITHIN 0.15
+REMARK 500 ANGSTROMS OF A SYMMETRY RELATED ATOM IS ASSUMED TO BE ON A
+REMARK 500 SPECIAL POSITION AND IS, THEREFORE, LISTED IN REMARK 375
+REMARK 500 INSTEAD OF REMARK 500. ATOMS WITH NON-BLANK ALTERNATE
+REMARK 500 LOCATION INDICATORS ARE NOT INCLUDED IN THE CALCULATIONS.
+REMARK 500
+REMARK 500 DISTANCE CUTOFF:
+REMARK 500 2.2 ANGSTROMS FOR CONTACTS NOT INVOLVING HYDROGEN ATOMS
+REMARK 500 1.6 ANGSTROMS FOR CONTACTS INVOLVING HYDROGEN ATOMS
+REMARK 500
+REMARK 500 ATM1 RES C SSEQI ATM2 RES C SSEQI SSYMOP DISTANCE
+REMARK 500 OD1 ASN A 19 ND2 ASN A 39 1556 2.09
+REMARK 500
+REMARK 500 REMARK: NULL
+REMARK 500
+REMARK 500 GEOMETRY AND STEREOCHEMISTRY
+REMARK 500 SUBTOPIC: COVALENT BOND ANGLES
+REMARK 500
+REMARK 500 THE STEREOCHEMICAL PARAMETERS OF THE FOLLOWING RESIDUES
+REMARK 500 HAVE VALUES WHICH DEVIATE FROM EXPECTED VALUES BY MORE
+REMARK 500 THAN 6*RMSD (M=MODEL NUMBER; RES=RESIDUE NAME; C=CHAIN
+REMARK 500 IDENTIFIER; SSEQ=SEQUENCE NUMBER; I=INSERTION CODE).
+REMARK 500
+REMARK 500 STANDARD TABLE:
+REMARK 500 FORMAT: (10X,I3,1X,A3,1X,A1,I4,A1,3(1X,A4,2X),12X,F5.1)
+REMARK 500
+REMARK 500 EXPECTED VALUES PROTEIN: ENGH AND HUBER, 1999
+REMARK 500 EXPECTED VALUES NUCLEIC ACID: CLOWNEY ET AL 1996
+REMARK 500
+REMARK 500 M RES CSSEQI ATM1 ATM2 ATM3
+REMARK 500 ARG A 14 NE - CZ - NH1 ANGL. DEV. = -4.2 DEGREES
+REMARK 500 ASP A 18 CB - CG - OD1 ANGL. DEV. = 6.1 DEGREES
+REMARK 500 ARG A 21 CD - NE - CZ ANGL. DEV. = 13.6 DEGREES
+REMARK 500 ARG A 21 NE - CZ - NH2 ANGL. DEV. = 5.1 DEGREES
+REMARK 500 ARG A 45 NE - CZ - NH1 ANGL. DEV. = 4.7 DEGREES
+REMARK 500 ARG A 45 NE - CZ - NH2 ANGL. DEV. = -5.7 DEGREES
+REMARK 500 ASP A 66 CB - CG - OD1 ANGL. DEV. = 6.6 DEGREES
+REMARK 500 ASP A 66 CB - CG - OD2 ANGL. DEV. = -7.2 DEGREES
+REMARK 500 ARG A 68 NE - CZ - NH1 ANGL. DEV. = 8.8 DEGREES
+REMARK 500 ARG A 68 NE - CZ - NH2 ANGL. DEV. = -6.5 DEGREES
+REMARK 500 ARG A 73 NE - CZ - NH2 ANGL. DEV. = -4.4 DEGREES
+REMARK 500 ASP A 87 CB - CG - OD1 ANGL. DEV. = 8.6 DEGREES
+REMARK 500 ARG A 112 NE - CZ - NH1 ANGL. DEV. = 4.4 DEGREES
+REMARK 500 ARG A 125 NE - CZ - NH2 ANGL. DEV. = -5.0 DEGREES
+REMARK 500 ARG A 128 NE - CZ - NH1 ANGL. DEV. = 3.0 DEGREES
+REMARK 500
+REMARK 500 REMARK: NULL
+REMARK 500
+REMARK 500 GEOMETRY AND STEREOCHEMISTRY
+REMARK 500 SUBTOPIC: PLANAR GROUPS
+REMARK 500
+REMARK 500 PLANAR GROUPS IN THE FOLLOWING RESIDUES HAVE A TOTAL
+REMARK 500 RMS DISTANCE OF ALL ATOMS FROM THE BEST-FIT PLANE
+REMARK 500 BY MORE THAN AN EXPECTED VALUE OF 6*RMSD, WITH AN
+REMARK 500 RMSD 0.02 ANGSTROMS, OR AT LEAST ONE ATOM HAS
+REMARK 500 AN RMSD GREATER THAN THIS VALUE
+REMARK 500 (M=MODEL NUMBER; RES=RESIDUE NAME; C=CHAIN IDENTIFIER;
+REMARK 500 SSEQ=SEQUENCE NUMBER; I=INSERTION CODE).
+REMARK 500
+REMARK 500 M RES CSSEQI RMS TYPE
+REMARK 500 ARG A 14 0.12 SIDE CHAIN
+REMARK 500 ARG A 21 0.21 SIDE CHAIN
+REMARK 500 ARG A 68 0.15 SIDE CHAIN
+REMARK 500 ARG A 73 0.25 SIDE CHAIN
+REMARK 500 ARG A 112 0.15 SIDE CHAIN
+REMARK 500 ARG A 114 0.13 SIDE CHAIN
+REMARK 500
+REMARK 500 REMARK: NULL
+REMARK 500
+REMARK 500 GEOMETRY AND STEREOCHEMISTRY
+REMARK 500 SUBTOPIC: MAIN CHAIN PLANARITY
+REMARK 500
+REMARK 500 THE FOLLOWING RESIDUES HAVE A PSEUDO PLANARITY
+REMARK 500 TORSION ANGLE, C(I) - CA(I) - N(I+1) - O(I), GREATER
+REMARK 500 10.0 DEGREES. (M=MODEL NUMBER; RES=RESIDUE NAME;
+REMARK 500 C=CHAIN IDENTIFIER; SSEQ=SEQUENCE NUMBER;
+REMARK 500 I=INSERTION CODE).
+REMARK 500
+REMARK 500 M RES CSSEQI ANGLE
+REMARK 500 ARG A 128 10.17
+REMARK 500
+REMARK 500 REMARK: NULL
+DBREF 1AKI A 1 129 UNP P00698 LYSC_CHICK 19 147
+SEQRES 1 A 129 LYS VAL PHE GLY ARG CYS GLU LEU ALA ALA ALA MET LYS
+SEQRES 2 A 129 ARG HIS GLY LEU ASP ASN TYR ARG GLY TYR SER LEU GLY
+SEQRES 3 A 129 ASN TRP VAL CYS ALA ALA LYS PHE GLU SER ASN PHE ASN
+SEQRES 4 A 129 THR GLN ALA THR ASN ARG ASN THR ASP GLY SER THR ASP
+SEQRES 5 A 129 TYR GLY ILE LEU GLN ILE ASN SER ARG TRP TRP CYS ASN
+SEQRES 6 A 129 ASP GLY ARG THR PRO GLY SER ARG ASN LEU CYS ASN ILE
+SEQRES 7 A 129 PRO CYS SER ALA LEU LEU SER SER ASP ILE THR ALA SER
+SEQRES 8 A 129 VAL ASN CYS ALA LYS LYS ILE VAL SER ASP GLY ASN GLY
+SEQRES 9 A 129 MET ASN ALA TRP VAL ALA TRP ARG ASN ARG CYS LYS GLY
+SEQRES 10 A 129 THR ASP VAL GLN ALA TRP ILE ARG GLY CYS ARG LEU
+FORMUL 2 HOH *78(H2 O)
+HELIX 1 1 ARG A 5 ARG A 14 1 10
+HELIX 2 2 TYR A 20 GLY A 22 5 3
+HELIX 3 3 LEU A 25 SER A 36 1 12
+HELIX 4 4 CYS A 80 LEU A 84 5 5
+HELIX 5 5 THR A 89 ASP A 101 1 13
+HELIX 6 6 GLY A 104 ALA A 107 5 4
+HELIX 7 7 VAL A 109 ARG A 114 1 6
+HELIX 8 8 VAL A 120 TRP A 123 5 4
+SHEET 1 A 2 THR A 43 ARG A 45 0
+SHEET 2 A 2 THR A 51 TYR A 53 -1 N ASP A 52 O ASN A 44
+SSBOND 1 CYS A 6 CYS A 127 1555 1555 1.97
+SSBOND 2 CYS A 30 CYS A 115 1555 1555 2.00
+SSBOND 3 CYS A 64 CYS A 80 1555 1555 1.99
+SSBOND 4 CYS A 76 CYS A 94 1555 1555 2.02
+CRYST1 59.062 68.451 30.517 90.00 90.00 90.00 P 21 21 21 4
+ORIGX1 1.000000 0.000000 0.000000 0.00000
+ORIGX2 0.000000 1.000000 0.000000 0.00000
+ORIGX3 0.000000 0.000000 1.000000 0.00000
+SCALE1 0.016931 0.000000 0.000000 0.00000
+SCALE2 0.000000 0.014609 0.000000 0.00000
+SCALE3 0.000000 0.000000 0.032769 0.00000
+ATOM 1 N LYS A 1 35.365 22.342 -11.980 1.00 22.28 N
+ATOM 2 CA LYS A 1 35.892 21.073 -11.427 1.00 21.12 C
+ATOM 3 C LYS A 1 34.741 20.264 -10.844 1.00 16.85 C
+ATOM 4 O LYS A 1 33.945 20.813 -10.081 1.00 18.94 O
+ATOM 5 CB LYS A 1 36.872 21.435 -10.306 1.00 20.78 C
+ATOM 6 CG LYS A 1 37.453 20.248 -9.565 1.00 18.47 C
+ATOM 7 CD LYS A 1 38.688 20.649 -8.775 1.00 20.32 C
+ATOM 8 CE LYS A 1 39.057 19.508 -7.837 1.00 24.76 C
+ATOM 9 NZ LYS A 1 40.423 19.771 -7.299 1.00 28.27 N
+ATOM 10 N VAL A 2 34.739 18.961 -11.042 1.00 19.96 N
+ATOM 11 CA VAL A 2 33.903 17.998 -10.333 1.00 18.10 C
+ATOM 12 C VAL A 2 34.800 17.312 -9.294 1.00 19.39 C
+ATOM 13 O VAL A 2 35.759 16.605 -9.665 1.00 22.14 O
+ATOM 14 CB VAL A 2 33.140 17.034 -11.232 1.00 16.81 C
+ATOM 15 CG1 VAL A 2 32.251 16.084 -10.434 1.00 21.92 C
+ATOM 16 CG2 VAL A 2 32.294 17.714 -12.290 1.00 19.46 C
+ATOM 17 N PHE A 3 34.491 17.546 -8.038 1.00 19.89 N
+ATOM 18 CA PHE A 3 35.185 16.903 -6.918 1.00 17.43 C
+ATOM 19 C PHE A 3 34.742 15.441 -6.771 1.00 15.70 C
+ATOM 20 O PHE A 3 33.525 15.162 -6.862 1.00 18.52 O
+ATOM 21 CB PHE A 3 34.967 17.632 -5.594 1.00 17.94 C
+ATOM 22 CG PHE A 3 35.944 18.737 -5.375 1.00 16.78 C
+ATOM 23 CD1 PHE A 3 35.666 20.050 -5.798 1.00 15.97 C
+ATOM 24 CD2 PHE A 3 37.000 18.557 -4.473 1.00 19.95 C
+ATOM 25 CE1 PHE A 3 36.577 21.076 -5.568 1.00 17.32 C
+ATOM 26 CE2 PHE A 3 37.869 19.589 -4.157 1.00 17.65 C
+ATOM 27 CZ PHE A 3 37.636 20.873 -4.666 1.00 17.91 C
+ATOM 28 N GLY A 4 35.724 14.639 -6.331 1.00 16.79 N
+ATOM 29 CA GLY A 4 35.366 13.280 -5.870 1.00 16.34 C
+ATOM 30 C GLY A 4 34.924 13.420 -4.415 1.00 11.91 C
+ATOM 31 O GLY A 4 35.303 14.403 -3.781 1.00 16.23 O
+ATOM 32 N ARG A 5 34.053 12.538 -3.973 1.00 14.65 N
+ATOM 33 CA ARG A 5 33.565 12.538 -2.588 1.00 15.91 C
+ATOM 34 C ARG A 5 34.665 12.734 -1.556 1.00 15.38 C
+ATOM 35 O ARG A 5 34.669 13.651 -0.704 1.00 13.15 O
+ATOM 36 CB ARG A 5 32.765 11.262 -2.331 1.00 17.38 C
+ATOM 37 CG ARG A 5 32.213 11.203 -0.920 1.00 13.79 C
+ATOM 38 CD ARG A 5 31.375 10.001 -0.722 1.00 15.84 C
+ATOM 39 NE ARG A 5 32.059 8.749 -0.958 1.00 18.74 N
+ATOM 40 CZ ARG A 5 32.733 8.011 -0.097 1.00 15.19 C
+ATOM 41 NH1 ARG A 5 32.836 8.332 1.187 1.00 17.50 N
+ATOM 42 NH2 ARG A 5 33.245 6.836 -0.526 1.00 23.44 N
+ATOM 43 N CYS A 6 35.674 11.853 -1.612 1.00 14.07 N
+ATOM 44 CA CYS A 6 36.781 11.870 -0.654 1.00 14.62 C
+ATOM 45 C CYS A 6 37.747 13.050 -0.777 1.00 10.99 C
+ATOM 46 O CYS A 6 38.148 13.609 0.264 1.00 16.34 O
+ATOM 47 CB CYS A 6 37.491 10.532 -0.621 1.00 16.90 C
+ATOM 48 SG CYS A 6 36.540 9.205 0.140 1.00 18.61 S
+ATOM 49 N GLU A 7 37.861 13.481 -2.019 1.00 14.24 N
+ATOM 50 CA GLU A 7 38.685 14.686 -2.311 1.00 13.83 C
+ATOM 51 C GLU A 7 38.049 15.926 -1.658 1.00 14.86 C
+ATOM 52 O GLU A 7 38.744 16.729 -1.011 1.00 15.01 O
+ATOM 53 CB GLU A 7 38.784 14.846 -3.818 1.00 14.85 C
+ATOM 54 CG GLU A 7 39.540 16.051 -4.379 1.00 18.50 C
+ATOM 55 CD GLU A 7 39.576 16.242 -5.870 1.00 20.16 C
+ATOM 56 OE1 GLU A 7 38.672 15.644 -6.491 1.00 26.20 O
+ATOM 57 OE2 GLU A 7 40.415 16.953 -6.381 1.00 25.49 O
+ATOM 58 N LEU A 8 36.743 16.049 -1.819 1.00 15.18 N
+ATOM 59 CA LEU A 8 35.964 17.158 -1.255 1.00 12.72 C
+ATOM 60 C LEU A 8 36.051 17.132 0.266 1.00 9.45 C
+ATOM 61 O LEU A 8 36.159 18.166 0.920 1.00 13.45 O
+ATOM 62 CB LEU A 8 34.528 17.172 -1.811 1.00 14.31 C
+ATOM 63 CG LEU A 8 33.718 18.354 -1.305 1.00 15.95 C
+ATOM 64 CD1 LEU A 8 34.297 19.656 -1.841 1.00 16.56 C
+ATOM 65 CD2 LEU A 8 32.246 18.143 -1.596 1.00 15.88 C
+ATOM 66 N ALA A 9 35.754 15.980 0.828 1.00 14.24 N
+ATOM 67 CA ALA A 9 35.838 15.757 2.284 1.00 13.25 C
+ATOM 68 C ALA A 9 37.144 16.314 2.840 1.00 12.89 C
+ATOM 69 O ALA A 9 37.151 16.978 3.897 1.00 14.78 O
+ATOM 70 CB ALA A 9 35.656 14.287 2.623 1.00 13.89 C
+ATOM 71 N ALA A 10 38.272 15.983 2.204 1.00 12.54 N
+ATOM 72 CA ALA A 10 39.610 16.431 2.616 1.00 16.58 C
+ATOM 73 C ALA A 10 39.736 17.944 2.459 1.00 15.35 C
+ATOM 74 O ALA A 10 40.193 18.499 3.469 1.00 17.40 O
+ATOM 75 CB ALA A 10 40.708 15.706 1.842 1.00 15.49 C
+ATOM 76 N ALA A 11 39.227 18.519 1.385 1.00 13.54 N
+ATOM 77 CA ALA A 11 39.264 19.982 1.223 1.00 15.23 C
+ATOM 78 C ALA A 11 38.491 20.702 2.321 1.00 15.68 C
+ATOM 79 O ALA A 11 38.946 21.658 2.953 1.00 16.87 O
+ATOM 80 CB ALA A 11 38.869 20.421 -0.175 1.00 14.12 C
+ATOM 81 N MET A 12 37.288 20.214 2.590 1.00 15.47 N
+ATOM 82 CA MET A 12 36.398 20.781 3.612 1.00 13.69 C
+ATOM 83 C MET A 12 36.990 20.715 5.007 1.00 12.55 C
+ATOM 84 O MET A 12 36.906 21.637 5.840 1.00 17.69 O
+ATOM 85 CB MET A 12 34.993 20.213 3.515 1.00 11.18 C
+ATOM 86 CG MET A 12 34.320 20.724 2.265 1.00 15.06 C
+ATOM 87 SD MET A 12 32.634 19.986 2.235 1.00 17.81 S
+ATOM 88 CE MET A 12 31.788 21.135 1.138 1.00 18.08 C
+ATOM 89 N LYS A 13 37.688 19.628 5.314 1.00 12.42 N
+ATOM 90 CA LYS A 13 38.387 19.385 6.579 1.00 14.58 C
+ATOM 91 C LYS A 13 39.460 20.467 6.731 1.00 14.55 C
+ATOM 92 O LYS A 13 39.507 21.137 7.776 1.00 16.49 O
+ATOM 93 CB LYS A 13 38.934 17.952 6.572 1.00 15.36 C
+ATOM 94 CG LYS A 13 39.742 17.555 7.798 1.00 20.46 C
+ATOM 95 CD LYS A 13 38.973 16.777 8.834 1.00 23.53 C
+ATOM 96 CE LYS A 13 39.293 15.305 8.751 1.00 26.37 C
+ATOM 97 NZ LYS A 13 38.077 14.461 8.946 1.00 30.88 N
+ATOM 98 N ARG A 14 40.267 20.629 5.688 1.00 18.91 N
+ATOM 99 CA ARG A 14 41.387 21.577 5.713 1.00 17.66 C
+ATOM 100 C ARG A 14 40.927 23.017 5.881 1.00 16.78 C
+ATOM 101 O ARG A 14 41.557 23.834 6.584 1.00 20.06 O
+ATOM 102 CB ARG A 14 42.388 21.351 4.601 1.00 20.89 C
+ATOM 103 CG ARG A 14 42.173 22.079 3.289 1.00 25.07 C
+ATOM 104 CD ARG A 14 43.444 22.075 2.490 1.00 23.98 C
+ATOM 105 NE ARG A 14 43.687 20.710 2.012 1.00 31.92 N
+ATOM 106 CZ ARG A 14 43.098 20.255 0.892 1.00 26.04 C
+ATOM 107 NH1 ARG A 14 42.695 21.186 0.018 1.00 33.46 N
+ATOM 108 NH2 ARG A 14 42.949 18.957 0.689 1.00 25.91 N
+ATOM 109 N HIS A 15 39.681 23.247 5.463 1.00 17.84 N
+ATOM 110 CA HIS A 15 39.075 24.591 5.526 1.00 15.99 C
+ATOM 111 C HIS A 15 38.310 24.861 6.814 1.00 17.70 C
+ATOM 112 O HIS A 15 37.608 25.875 6.952 1.00 22.68 O
+ATOM 113 CB HIS A 15 38.223 24.918 4.285 1.00 19.19 C
+ATOM 114 CG HIS A 15 39.085 25.388 3.171 1.00 20.14 C
+ATOM 115 ND1 HIS A 15 39.457 24.635 2.091 1.00 24.53 N
+ATOM 116 CD2 HIS A 15 39.739 26.570 2.993 1.00 24.44 C
+ATOM 117 CE1 HIS A 15 40.211 25.312 1.258 1.00 20.68 C
+ATOM 118 NE2 HIS A 15 40.524 26.419 1.889 1.00 27.34 N
+ATOM 119 N GLY A 16 38.296 23.927 7.720 1.00 17.50 N
+ATOM 120 CA GLY A 16 37.765 24.025 9.072 1.00 18.71 C
+ATOM 121 C GLY A 16 36.264 23.799 9.210 1.00 18.40 C
+ATOM 122 O GLY A 16 35.646 24.400 10.127 1.00 21.57 O
+ATOM 123 N LEU A 17 35.665 23.073 8.274 1.00 17.56 N
+ATOM 124 CA LEU A 17 34.238 22.795 8.298 1.00 15.38 C
+ATOM 125 C LEU A 17 33.845 21.682 9.266 1.00 18.28 C
+ATOM 126 O LEU A 17 32.643 21.594 9.552 1.00 18.24 O
+ATOM 127 CB LEU A 17 33.648 22.647 6.901 1.00 15.02 C
+ATOM 128 CG LEU A 17 33.451 23.889 6.060 1.00 16.08 C
+ATOM 129 CD1 LEU A 17 32.933 23.420 4.705 1.00 13.83 C
+ATOM 130 CD2 LEU A 17 32.556 24.946 6.679 1.00 18.60 C
+ATOM 131 N ASP A 18 34.785 20.814 9.605 1.00 16.36 N
+ATOM 132 CA ASP A 18 34.492 19.722 10.526 1.00 16.25 C
+ATOM 133 C ASP A 18 34.015 20.222 11.901 1.00 17.32 C
+ATOM 134 O ASP A 18 34.826 20.778 12.658 1.00 20.27 O
+ATOM 135 CB ASP A 18 35.557 18.633 10.598 1.00 19.37 C
+ATOM 136 CG ASP A 18 35.017 17.408 11.311 1.00 20.25 C
+ATOM 137 OD1 ASP A 18 33.805 17.154 11.455 1.00 21.00 O
+ATOM 138 OD2 ASP A 18 35.925 16.603 11.662 1.00 27.06 O
+ATOM 139 N ASN A 19 32.746 19.990 12.205 1.00 18.55 N
+ATOM 140 CA ASN A 19 32.123 20.431 13.448 1.00 17.19 C
+ATOM 141 C ASN A 19 31.854 21.941 13.497 1.00 16.61 C
+ATOM 142 O ASN A 19 31.426 22.398 14.573 1.00 18.56 O
+ATOM 143 CB ASN A 19 32.767 20.004 14.770 1.00 18.92 C
+ATOM 144 CG ASN A 19 32.162 20.512 16.064 1.00 24.64 C
+ATOM 145 OD1 ASN A 19 30.967 20.273 16.355 1.00 32.53 O
+ATOM 146 ND2 ASN A 19 32.847 21.361 16.852 1.00 24.14 N
+ATOM 147 N TYR A 20 31.969 22.650 12.406 1.00 16.84 N
+ATOM 148 CA TYR A 20 31.707 24.099 12.411 1.00 15.54 C
+ATOM 149 C TYR A 20 30.231 24.343 12.759 1.00 15.81 C
+ATOM 150 O TYR A 20 29.288 23.874 12.118 1.00 16.89 O
+ATOM 151 CB TYR A 20 32.070 24.736 11.066 1.00 18.16 C
+ATOM 152 CG TYR A 20 32.061 26.250 11.144 1.00 20.28 C
+ATOM 153 CD1 TYR A 20 33.141 26.886 11.760 1.00 21.42 C
+ATOM 154 CD2 TYR A 20 30.979 27.004 10.691 1.00 20.44 C
+ATOM 155 CE1 TYR A 20 33.206 28.277 11.794 1.00 22.24 C
+ATOM 156 CE2 TYR A 20 31.018 28.399 10.779 1.00 21.36 C
+ATOM 157 CZ TYR A 20 32.102 29.017 11.383 1.00 19.38 C
+ATOM 158 OH TYR A 20 32.136 30.371 11.525 1.00 26.77 O
+ATOM 159 N ARG A 21 30.088 25.142 13.803 1.00 18.43 N
+ATOM 160 CA ARG A 21 28.774 25.553 14.319 1.00 15.68 C
+ATOM 161 C ARG A 21 27.964 24.312 14.623 1.00 15.17 C
+ATOM 162 O ARG A 21 26.733 24.333 14.606 1.00 18.36 O
+ATOM 163 CB ARG A 21 28.014 26.565 13.453 1.00 19.21 C
+ATOM 164 CG ARG A 21 28.507 27.995 13.535 1.00 21.50 C
+ATOM 165 CD ARG A 21 28.110 28.755 14.765 1.00 25.00 C
+ATOM 166 NE ARG A 21 29.319 29.321 15.324 1.00 30.37 N
+ATOM 167 CZ ARG A 21 30.215 30.234 14.978 1.00 29.02 C
+ATOM 168 NH1 ARG A 21 31.501 29.856 14.846 1.00 29.04 N
+ATOM 169 NH2 ARG A 21 29.938 31.437 14.495 1.00 31.89 N
+ATOM 170 N GLY A 22 28.689 23.250 14.998 1.00 17.81 N
+ATOM 171 CA GLY A 22 28.103 22.021 15.519 1.00 17.72 C
+ATOM 172 C GLY A 22 27.748 21.018 14.436 1.00 18.89 C
+ATOM 173 O GLY A 22 27.085 20.022 14.784 1.00 23.26 O
+ATOM 174 N TYR A 23 28.209 21.230 13.216 1.00 18.20 N
+ATOM 175 CA TYR A 23 27.887 20.318 12.111 1.00 15.42 C
+ATOM 176 C TYR A 23 29.124 19.533 11.628 1.00 16.30 C
+ATOM 177 O TYR A 23 30.045 20.191 11.139 1.00 16.66 O
+ATOM 178 CB TYR A 23 27.351 21.107 10.913 1.00 15.82 C
+ATOM 179 CG TYR A 23 26.001 21.745 11.127 1.00 15.96 C
+ATOM 180 CD1 TYR A 23 24.846 20.962 11.139 1.00 14.81 C
+ATOM 181 CD2 TYR A 23 25.897 23.096 11.458 1.00 16.60 C
+ATOM 182 CE1 TYR A 23 23.600 21.518 11.422 1.00 17.08 C
+ATOM 183 CE2 TYR A 23 24.647 23.673 11.726 1.00 19.34 C
+ATOM 184 CZ TYR A 23 23.518 22.881 11.701 1.00 19.21 C
+ATOM 185 OH TYR A 23 22.289 23.438 11.912 1.00 25.61 O
+ATOM 186 N SER A 24 29.029 18.223 11.810 1.00 15.35 N
+ATOM 187 CA SER A 24 30.143 17.347 11.414 1.00 16.89 C
+ATOM 188 C SER A 24 30.359 17.379 9.895 1.00 15.61 C
+ATOM 189 O SER A 24 29.442 17.687 9.139 1.00 13.88 O
+ATOM 190 CB SER A 24 29.922 15.934 11.907 1.00 17.54 C
+ATOM 191 OG SER A 24 28.799 15.336 11.308 1.00 19.85 O
+ATOM 192 N LEU A 25 31.593 17.028 9.540 1.00 16.08 N
+ATOM 193 CA LEU A 25 32.035 17.092 8.138 1.00 13.16 C
+ATOM 194 C LEU A 25 31.030 16.437 7.183 1.00 13.39 C
+ATOM 195 O LEU A 25 30.874 16.924 6.056 1.00 15.34 O
+ATOM 196 CB LEU A 25 33.410 16.409 8.084 1.00 13.47 C
+ATOM 197 CG LEU A 25 34.015 16.477 6.689 1.00 12.91 C
+ATOM 198 CD1 LEU A 25 34.174 17.929 6.289 1.00 13.04 C
+ATOM 199 CD2 LEU A 25 35.398 15.810 6.752 1.00 14.54 C
+ATOM 200 N GLY A 26 30.501 15.280 7.576 1.00 13.10 N
+ATOM 201 CA GLY A 26 29.539 14.561 6.756 1.00 13.88 C
+ATOM 202 C GLY A 26 28.338 15.378 6.277 1.00 12.12 C
+ATOM 203 O GLY A 26 27.886 15.250 5.120 1.00 13.13 O
+ATOM 204 N ASN A 27 27.905 16.281 7.161 1.00 12.16 N
+ATOM 205 CA ASN A 27 26.827 17.227 6.857 1.00 14.74 C
+ATOM 206 C ASN A 27 27.164 18.015 5.597 1.00 15.43 C
+ATOM 207 O ASN A 27 26.317 18.208 4.720 1.00 15.27 O
+ATOM 208 CB ASN A 27 26.484 18.126 8.054 1.00 12.34 C
+ATOM 209 CG ASN A 27 25.681 17.267 9.048 1.00 12.22 C
+ATOM 210 OD1 ASN A 27 24.511 16.933 8.820 1.00 17.40 O
+ATOM 211 ND2 ASN A 27 26.348 17.052 10.169 1.00 18.27 N
+ATOM 212 N TRP A 28 28.344 18.591 5.583 1.00 16.78 N
+ATOM 213 CA TRP A 28 28.831 19.475 4.513 1.00 16.49 C
+ATOM 214 C TRP A 28 28.940 18.741 3.183 1.00 12.99 C
+ATOM 215 O TRP A 28 28.742 19.321 2.090 1.00 13.97 O
+ATOM 216 CB TRP A 28 30.104 20.145 5.023 1.00 14.55 C
+ATOM 217 CG TRP A 28 29.941 20.978 6.251 1.00 11.93 C
+ATOM 218 CD1 TRP A 28 30.176 20.624 7.546 1.00 14.42 C
+ATOM 219 CD2 TRP A 28 29.319 22.284 6.287 1.00 13.11 C
+ATOM 220 NE1 TRP A 28 29.924 21.690 8.365 1.00 16.94 N
+ATOM 221 CE2 TRP A 28 29.337 22.679 7.641 1.00 14.07 C
+ATOM 222 CE3 TRP A 28 28.894 23.168 5.295 1.00 15.97 C
+ATOM 223 CZ2 TRP A 28 28.913 23.943 8.038 1.00 18.07 C
+ATOM 224 CZ3 TRP A 28 28.398 24.404 5.682 1.00 18.26 C
+ATOM 225 CH2 TRP A 28 28.431 24.766 7.025 1.00 17.18 C
+ATOM 226 N VAL A 29 29.572 17.543 3.261 1.00 13.00 N
+ATOM 227 CA VAL A 29 29.729 16.711 2.047 1.00 14.15 C
+ATOM 228 C VAL A 29 28.379 16.340 1.429 1.00 10.43 C
+ATOM 229 O VAL A 29 28.166 16.496 0.228 1.00 13.40 O
+ATOM 230 CB VAL A 29 30.649 15.492 2.359 1.00 12.76 C
+ATOM 231 CG1 VAL A 29 30.782 14.596 1.136 1.00 15.37 C
+ATOM 232 CG2 VAL A 29 32.010 16.050 2.772 1.00 14.18 C
+ATOM 233 N CYS A 30 27.501 15.906 2.299 1.00 15.32 N
+ATOM 234 CA CYS A 30 26.115 15.567 1.991 1.00 15.59 C
+ATOM 235 C CYS A 30 25.388 16.723 1.302 1.00 12.37 C
+ATOM 236 O CYS A 30 24.894 16.516 0.172 1.00 14.44 O
+ATOM 237 CB CYS A 30 25.343 15.046 3.172 1.00 14.99 C
+ATOM 238 SG CYS A 30 23.719 14.376 2.695 1.00 17.71 S
+ATOM 239 N ALA A 31 25.533 17.913 1.870 1.00 16.06 N
+ATOM 240 CA ALA A 31 24.949 19.130 1.305 1.00 15.82 C
+ATOM 241 C ALA A 31 25.487 19.354 -0.115 1.00 15.34 C
+ATOM 242 O ALA A 31 24.675 19.686 -0.998 1.00 16.00 O
+ATOM 243 CB ALA A 31 25.167 20.335 2.200 1.00 14.90 C
+ATOM 244 N ALA A 32 26.807 19.354 -0.286 1.00 11.93 N
+ATOM 245 CA ALA A 32 27.461 19.536 -1.579 1.00 13.08 C
+ATOM 246 C ALA A 32 26.943 18.538 -2.620 1.00 12.38 C
+ATOM 247 O ALA A 32 26.767 18.857 -3.789 1.00 13.89 O
+ATOM 248 CB ALA A 32 28.982 19.476 -1.398 1.00 14.17 C
+ATOM 249 N LYS A 33 26.731 17.303 -2.193 1.00 15.16 N
+ATOM 250 CA LYS A 33 26.261 16.216 -3.037 1.00 16.16 C
+ATOM 251 C LYS A 33 24.903 16.555 -3.658 1.00 15.04 C
+ATOM 252 O LYS A 33 24.806 16.511 -4.890 1.00 15.00 O
+ATOM 253 CB LYS A 33 26.221 14.860 -2.351 1.00 16.71 C
+ATOM 254 CG LYS A 33 25.697 13.696 -3.185 1.00 19.51 C
+ATOM 255 CD LYS A 33 26.498 13.400 -4.446 1.00 17.12 C
+ATOM 256 CE LYS A 33 25.686 12.464 -5.331 1.00 24.06 C
+ATOM 257 NZ LYS A 33 26.423 11.979 -6.525 1.00 26.78 N
+ATOM 258 N PHE A 34 23.972 16.946 -2.817 1.00 16.81 N
+ATOM 259 CA PHE A 34 22.569 17.125 -3.247 1.00 17.51 C
+ATOM 260 C PHE A 34 22.346 18.506 -3.836 1.00 18.19 C
+ATOM 261 O PHE A 34 21.504 18.692 -4.759 1.00 20.21 O
+ATOM 262 CB PHE A 34 21.626 16.673 -2.130 1.00 18.58 C
+ATOM 263 CG PHE A 34 21.644 15.172 -1.965 1.00 22.22 C
+ATOM 264 CD1 PHE A 34 21.209 14.353 -3.007 1.00 20.24 C
+ATOM 265 CD2 PHE A 34 22.272 14.627 -0.851 1.00 20.00 C
+ATOM 266 CE1 PHE A 34 21.372 12.961 -2.910 1.00 22.03 C
+ATOM 267 CE2 PHE A 34 22.443 13.245 -0.743 1.00 21.96 C
+ATOM 268 CZ PHE A 34 21.923 12.406 -1.737 1.00 20.77 C
+ATOM 269 N GLU A 35 23.251 19.415 -3.451 1.00 15.27 N
+ATOM 270 CA GLU A 35 23.178 20.789 -3.996 1.00 15.45 C
+ATOM 271 C GLU A 35 23.661 20.944 -5.423 1.00 17.45 C
+ATOM 272 O GLU A 35 23.014 21.514 -6.310 1.00 17.91 O
+ATOM 273 CB GLU A 35 23.698 21.892 -3.107 1.00 14.42 C
+ATOM 274 CG GLU A 35 22.994 22.212 -1.809 1.00 11.08 C
+ATOM 275 CD GLU A 35 21.631 22.846 -1.864 1.00 13.55 C
+ATOM 276 OE1 GLU A 35 21.408 23.360 -2.981 1.00 20.77 O
+ATOM 277 OE2 GLU A 35 20.947 23.032 -0.874 1.00 22.72 O
+ATOM 278 N SER A 36 24.867 20.483 -5.674 1.00 15.66 N
+ATOM 279 CA SER A 36 25.626 20.615 -6.903 1.00 17.79 C
+ATOM 280 C SER A 36 26.139 19.341 -7.569 1.00 16.97 C
+ATOM 281 O SER A 36 26.750 19.464 -8.642 1.00 21.78 O
+ATOM 282 CB SER A 36 26.830 21.528 -6.654 1.00 18.12 C
+ATOM 283 OG SER A 36 27.747 20.951 -5.748 1.00 15.16 O
+ATOM 284 N ASN A 37 25.957 18.222 -6.927 1.00 20.15 N
+ATOM 285 CA ASN A 37 26.628 16.968 -7.297 1.00 20.85 C
+ATOM 286 C ASN A 37 28.149 17.091 -7.302 1.00 19.72 C
+ATOM 287 O ASN A 37 28.813 16.627 -8.254 1.00 20.79 O
+ATOM 288 CB ASN A 37 26.028 16.490 -8.617 1.00 20.82 C
+ATOM 289 CG ASN A 37 26.156 14.980 -8.782 1.00 23.63 C
+ATOM 290 OD1 ASN A 37 26.640 14.266 -7.885 1.00 28.05 O
+ATOM 291 ND2 ASN A 37 25.867 14.506 -9.990 1.00 29.61 N
+ATOM 292 N PHE A 38 28.691 17.882 -6.383 1.00 16.12 N
+ATOM 293 CA PHE A 38 30.129 18.115 -6.279 1.00 14.75 C
+ATOM 294 C PHE A 38 30.717 18.903 -7.448 1.00 13.86 C
+ATOM 295 O PHE A 38 31.923 18.794 -7.702 1.00 16.66 O
+ATOM 296 CB PHE A 38 30.914 16.823 -6.047 1.00 17.09 C
+ATOM 297 CG PHE A 38 30.487 16.006 -4.863 1.00 15.96 C
+ATOM 298 CD1 PHE A 38 30.100 16.572 -3.655 1.00 15.43 C
+ATOM 299 CD2 PHE A 38 30.507 14.603 -4.993 1.00 17.66 C
+ATOM 300 CE1 PHE A 38 29.766 15.808 -2.558 1.00 16.83 C
+ATOM 301 CE2 PHE A 38 30.136 13.814 -3.891 1.00 15.86 C
+ATOM 302 CZ PHE A 38 29.835 14.410 -2.646 1.00 18.90 C
+ATOM 303 N ASN A 39 29.936 19.792 -8.033 1.00 17.63 N
+ATOM 304 CA ASN A 39 30.341 20.573 -9.199 1.00 16.77 C
+ATOM 305 C ASN A 39 30.543 22.034 -8.823 1.00 17.13 C
+ATOM 306 O ASN A 39 29.544 22.649 -8.423 1.00 17.24 O
+ATOM 307 CB ASN A 39 29.406 20.300 -10.366 1.00 17.40 C
+ATOM 308 CG ASN A 39 29.876 20.771 -11.716 1.00 16.96 C
+ATOM 309 OD1 ASN A 39 30.579 21.750 -11.956 1.00 23.22 O
+ATOM 310 ND2 ASN A 39 29.578 19.864 -12.653 1.00 26.35 N
+ATOM 311 N THR A 40 31.766 22.492 -8.928 1.00 14.88 N
+ATOM 312 CA THR A 40 32.139 23.869 -8.657 1.00 16.08 C
+ATOM 313 C THR A 40 31.504 24.890 -9.589 1.00 16.31 C
+ATOM 314 O THR A 40 31.316 26.054 -9.213 1.00 17.50 O
+ATOM 315 CB THR A 40 33.684 24.103 -8.422 1.00 17.07 C
+ATOM 316 OG1 THR A 40 34.270 24.156 -9.775 1.00 23.76 O
+ATOM 317 CG2 THR A 40 34.414 23.119 -7.491 1.00 16.46 C
+ATOM 318 N GLN A 41 31.001 24.430 -10.706 1.00 16.39 N
+ATOM 319 CA GLN A 41 30.524 25.291 -11.812 1.00 16.67 C
+ATOM 320 C GLN A 41 28.993 25.381 -11.874 1.00 15.44 C
+ATOM 321 O GLN A 41 28.504 26.019 -12.837 1.00 18.55 O
+ATOM 322 CB GLN A 41 31.047 24.817 -13.168 1.00 19.65 C
+ATOM 323 CG GLN A 41 32.549 25.004 -13.279 1.00 21.26 C
+ATOM 324 CD GLN A 41 32.763 26.236 -14.129 1.00 26.23 C
+ATOM 325 OE1 GLN A 41 32.356 26.308 -15.291 1.00 24.68 O
+ATOM 326 NE2 GLN A 41 33.276 27.231 -13.420 1.00 25.96 N
+ATOM 327 N ALA A 42 28.329 24.640 -11.012 1.00 15.95 N
+ATOM 328 CA ALA A 42 26.859 24.579 -11.061 1.00 17.42 C
+ATOM 329 C ALA A 42 26.257 25.969 -10.807 1.00 18.93 C
+ATOM 330 O ALA A 42 26.645 26.686 -9.884 1.00 17.00 O
+ATOM 331 CB ALA A 42 26.355 23.604 -9.998 1.00 22.79 C
+ATOM 332 N THR A 43 25.276 26.293 -11.643 1.00 18.30 N
+ATOM 333 CA THR A 43 24.441 27.490 -11.476 1.00 17.73 C
+ATOM 334 C THR A 43 22.976 27.112 -11.714 1.00 19.61 C
+ATOM 335 O THR A 43 22.715 26.271 -12.594 1.00 22.06 O
+ATOM 336 CB THR A 43 24.814 28.728 -12.375 1.00 17.58 C
+ATOM 337 OG1 THR A 43 24.555 28.321 -13.756 1.00 20.94 O
+ATOM 338 CG2 THR A 43 26.247 29.213 -12.184 1.00 18.26 C
+ATOM 339 N ASN A 44 22.088 27.742 -10.971 1.00 19.77 N
+ATOM 340 CA ASN A 44 20.640 27.497 -11.108 1.00 19.51 C
+ATOM 341 C ASN A 44 19.903 28.842 -10.963 1.00 14.85 C
+ATOM 342 O ASN A 44 20.169 29.600 -10.033 1.00 19.43 O
+ATOM 343 CB ASN A 44 20.155 26.480 -10.080 1.00 19.93 C
+ATOM 344 CG ASN A 44 18.646 26.315 -10.115 1.00 22.01 C
+ATOM 345 OD1 ASN A 44 18.128 25.720 -11.078 1.00 29.53 O
+ATOM 346 ND2 ASN A 44 17.906 26.927 -9.188 1.00 22.94 N
+ATOM 347 N ARG A 45 19.058 29.117 -11.924 1.00 16.90 N
+ATOM 348 CA ARG A 45 18.303 30.363 -12.015 1.00 18.37 C
+ATOM 349 C ARG A 45 16.955 30.163 -11.326 1.00 18.76 C
+ATOM 350 O ARG A 45 16.396 29.062 -11.425 1.00 20.87 O
+ATOM 351 CB ARG A 45 18.143 30.836 -13.462 1.00 19.30 C
+ATOM 352 CG ARG A 45 17.012 31.859 -13.557 1.00 23.98 C
+ATOM 353 CD ARG A 45 17.502 33.134 -12.930 1.00 23.36 C
+ATOM 354 NE ARG A 45 18.311 33.758 -13.981 1.00 29.56 N
+ATOM 355 CZ ARG A 45 17.620 34.271 -15.020 1.00 27.18 C
+ATOM 356 NH1 ARG A 45 16.287 34.331 -15.098 1.00 31.99 N
+ATOM 357 NH2 ARG A 45 18.374 34.698 -16.030 1.00 32.43 N
+ATOM 358 N ASN A 46 16.553 31.171 -10.554 1.00 18.13 N
+ATOM 359 CA ASN A 46 15.304 31.071 -9.782 1.00 20.10 C
+ATOM 360 C ASN A 46 14.261 32.063 -10.313 1.00 17.35 C
+ATOM 361 O ASN A 46 14.617 33.104 -10.880 1.00 19.15 O
+ATOM 362 CB ASN A 46 15.576 31.229 -8.295 1.00 20.05 C
+ATOM 363 CG ASN A 46 16.543 30.240 -7.679 1.00 20.12 C
+ATOM 364 OD1 ASN A 46 17.659 30.661 -7.346 1.00 21.21 O
+ATOM 365 ND2 ASN A 46 16.125 28.975 -7.600 1.00 19.44 N
+ATOM 366 N THR A 47 13.027 31.834 -9.887 1.00 19.96 N
+ATOM 367 CA THR A 47 11.871 32.654 -10.246 1.00 19.12 C
+ATOM 368 C THR A 47 12.001 34.107 -9.810 1.00 19.44 C
+ATOM 369 O THR A 47 11.600 34.969 -10.606 1.00 23.16 O
+ATOM 370 CB THR A 47 10.499 32.017 -9.789 1.00 17.70 C
+ATOM 371 OG1 THR A 47 10.507 32.195 -8.342 1.00 23.76 O
+ATOM 372 CG2 THR A 47 10.331 30.554 -10.188 1.00 22.66 C
+ATOM 373 N ASP A 48 12.625 34.377 -8.683 1.00 19.25 N
+ATOM 374 CA ASP A 48 12.885 35.716 -8.168 1.00 17.33 C
+ATOM 375 C ASP A 48 14.010 36.505 -8.823 1.00 17.71 C
+ATOM 376 O ASP A 48 14.246 37.621 -8.309 1.00 23.04 O
+ATOM 377 CB ASP A 48 13.023 35.735 -6.640 1.00 18.02 C
+ATOM 378 CG ASP A 48 14.367 35.174 -6.168 1.00 17.88 C
+ATOM 379 OD1 ASP A 48 15.104 34.602 -6.991 1.00 18.55 O
+ATOM 380 OD2 ASP A 48 14.750 35.442 -5.013 1.00 22.86 O
+ATOM 381 N GLY A 49 14.650 36.018 -9.862 1.00 17.62 N
+ATOM 382 CA GLY A 49 15.744 36.702 -10.546 1.00 17.27 C
+ATOM 383 C GLY A 49 17.147 36.315 -10.096 1.00 16.80 C
+ATOM 384 O GLY A 49 18.168 36.688 -10.694 1.00 20.27 O
+ATOM 385 N SER A 50 17.173 35.661 -8.930 1.00 19.61 N
+ATOM 386 CA SER A 50 18.435 35.225 -8.279 1.00 14.28 C
+ATOM 387 C SER A 50 18.977 33.963 -8.941 1.00 18.09 C
+ATOM 388 O SER A 50 18.273 33.255 -9.697 1.00 16.76 O
+ATOM 389 CB SER A 50 18.272 35.089 -6.781 1.00 17.64 C
+ATOM 390 OG SER A 50 17.530 33.930 -6.463 1.00 17.54 O
+ATOM 391 N THR A 51 20.271 33.766 -8.734 1.00 16.00 N
+ATOM 392 CA THR A 51 20.970 32.557 -9.202 1.00 13.82 C
+ATOM 393 C THR A 51 21.718 31.969 -8.000 1.00 14.58 C
+ATOM 394 O THR A 51 22.160 32.712 -7.119 1.00 13.28 O
+ATOM 395 CB THR A 51 21.904 32.908 -10.419 1.00 12.66 C
+ATOM 396 OG1 THR A 51 21.099 33.576 -11.407 1.00 17.65 O
+ATOM 397 CG2 THR A 51 22.686 31.699 -10.952 1.00 14.39 C
+ATOM 398 N ASP A 52 21.708 30.650 -7.927 1.00 14.66 N
+ATOM 399 CA ASP A 52 22.528 29.900 -6.959 1.00 13.43 C
+ATOM 400 C ASP A 52 23.843 29.488 -7.635 1.00 12.35 C
+ATOM 401 O ASP A 52 23.847 29.066 -8.805 1.00 15.18 O
+ATOM 402 CB ASP A 52 21.765 28.649 -6.554 1.00 14.12 C
+ATOM 403 CG ASP A 52 20.396 28.991 -6.002 1.00 20.03 C
+ATOM 404 OD1 ASP A 52 20.220 29.928 -5.237 1.00 21.30 O
+ATOM 405 OD2 ASP A 52 19.517 28.144 -6.222 1.00 20.92 O
+ATOM 406 N TYR A 53 24.924 29.734 -6.905 1.00 14.56 N
+ATOM 407 CA TYR A 53 26.278 29.604 -7.450 1.00 13.61 C
+ATOM 408 C TYR A 53 27.161 28.595 -6.708 1.00 12.70 C
+ATOM 409 O TYR A 53 27.289 28.606 -5.486 1.00 13.36 O
+ATOM 410 CB TYR A 53 26.993 30.972 -7.489 1.00 12.10 C
+ATOM 411 CG TYR A 53 26.437 31.959 -8.487 1.00 11.48 C
+ATOM 412 CD1 TYR A 53 26.843 32.003 -9.821 1.00 17.21 C
+ATOM 413 CD2 TYR A 53 25.510 32.907 -8.050 1.00 14.42 C
+ATOM 414 CE1 TYR A 53 26.291 32.922 -10.717 1.00 17.40 C
+ATOM 415 CE2 TYR A 53 24.907 33.803 -8.932 1.00 12.97 C
+ATOM 416 CZ TYR A 53 25.357 33.847 -10.252 1.00 15.91 C
+ATOM 417 OH TYR A 53 24.864 34.804 -11.094 1.00 19.31 O
+ATOM 418 N GLY A 54 27.751 27.721 -7.493 1.00 16.54 N
+ATOM 419 CA GLY A 54 28.845 26.832 -7.121 1.00 15.67 C
+ATOM 420 C GLY A 54 28.499 25.601 -6.290 1.00 10.78 C
+ATOM 421 O GLY A 54 27.339 25.155 -6.255 1.00 13.16 O
+ATOM 422 N ILE A 55 29.560 25.088 -5.684 1.00 15.43 N
+ATOM 423 CA ILE A 55 29.452 23.795 -4.994 1.00 14.01 C
+ATOM 424 C ILE A 55 28.424 23.748 -3.872 1.00 12.26 C
+ATOM 425 O ILE A 55 27.770 22.697 -3.693 1.00 16.92 O
+ATOM 426 CB ILE A 55 30.891 23.340 -4.563 1.00 16.64 C
+ATOM 427 CG1 ILE A 55 30.785 21.812 -4.319 1.00 17.00 C
+ATOM 428 CG2 ILE A 55 31.396 24.210 -3.390 1.00 18.87 C
+ATOM 429 CD1 ILE A 55 32.101 21.018 -4.339 1.00 20.60 C
+ATOM 430 N LEU A 56 28.216 24.881 -3.236 1.00 12.85 N
+ATOM 431 CA LEU A 56 27.173 24.998 -2.203 1.00 14.96 C
+ATOM 432 C LEU A 56 25.981 25.838 -2.659 1.00 11.08 C
+ATOM 433 O LEU A 56 25.161 26.111 -1.774 1.00 16.40 O
+ATOM 434 CB LEU A 56 27.816 25.424 -0.877 1.00 14.44 C
+ATOM 435 CG LEU A 56 28.692 24.348 -0.204 1.00 14.92 C
+ATOM 436 CD1 LEU A 56 29.331 24.997 1.008 1.00 18.78 C
+ATOM 437 CD2 LEU A 56 27.808 23.177 0.192 1.00 19.47 C
+ATOM 438 N GLN A 57 25.865 26.104 -3.946 1.00 14.17 N
+ATOM 439 CA GLN A 57 24.668 26.785 -4.465 1.00 11.55 C
+ATOM 440 C GLN A 57 24.277 27.940 -3.558 1.00 15.09 C
+ATOM 441 O GLN A 57 23.116 28.031 -3.094 1.00 15.28 O
+ATOM 442 CB GLN A 57 23.521 25.765 -4.549 1.00 13.56 C
+ATOM 443 CG GLN A 57 23.733 24.790 -5.699 1.00 12.45 C
+ATOM 444 CD GLN A 57 23.684 25.440 -7.069 1.00 14.21 C
+ATOM 445 OE1 GLN A 57 22.574 25.602 -7.591 1.00 18.18 O
+ATOM 446 NE2 GLN A 57 24.813 25.805 -7.651 1.00 14.86 N
+ATOM 447 N ILE A 58 25.164 28.910 -3.428 1.00 14.04 N
+ATOM 448 CA ILE A 58 24.930 30.118 -2.649 1.00 16.09 C
+ATOM 449 C ILE A 58 24.204 31.163 -3.504 1.00 11.19 C
+ATOM 450 O ILE A 58 24.555 31.405 -4.667 1.00 14.34 O
+ATOM 451 CB ILE A 58 26.301 30.665 -2.134 1.00 14.90 C
+ATOM 452 CG1 ILE A 58 26.751 29.724 -0.985 1.00 13.85 C
+ATOM 453 CG2 ILE A 58 26.178 32.135 -1.693 1.00 15.47 C
+ATOM 454 CD1 ILE A 58 28.246 29.954 -0.641 1.00 16.12 C
+ATOM 455 N ASN A 59 23.145 31.671 -2.905 1.00 14.23 N
+ATOM 456 CA ASN A 59 22.146 32.510 -3.590 1.00 10.60 C
+ATOM 457 C ASN A 59 22.550 33.974 -3.730 1.00 13.03 C
+ATOM 458 O ASN A 59 22.917 34.607 -2.740 1.00 18.49 O
+ATOM 459 CB ASN A 59 20.805 32.306 -2.884 1.00 15.76 C
+ATOM 460 CG ASN A 59 19.650 32.986 -3.588 1.00 15.71 C
+ATOM 461 OD1 ASN A 59 19.464 34.171 -3.244 1.00 18.79 O
+ATOM 462 ND2 ASN A 59 19.155 32.320 -4.612 1.00 16.44 N
+ATOM 463 N SER A 60 22.268 34.485 -4.941 1.00 11.71 N
+ATOM 464 CA SER A 60 22.570 35.910 -5.255 1.00 13.81 C
+ATOM 465 C SER A 60 21.687 36.993 -4.642 1.00 15.28 C
+ATOM 466 O SER A 60 22.054 38.177 -4.774 1.00 17.67 O
+ATOM 467 CB SER A 60 22.643 36.139 -6.750 1.00 14.62 C
+ATOM 468 OG SER A 60 21.432 36.007 -7.444 1.00 14.30 O
+ATOM 469 N ARG A 61 20.526 36.660 -4.138 1.00 15.88 N
+ATOM 470 CA ARG A 61 19.691 37.653 -3.434 1.00 18.54 C
+ATOM 471 C ARG A 61 20.277 38.108 -2.106 1.00 19.97 C
+ATOM 472 O ARG A 61 20.282 39.333 -1.876 1.00 26.30 O
+ATOM 473 CB ARG A 61 18.267 37.195 -3.250 1.00 18.00 C
+ATOM 474 CG ARG A 61 17.315 38.350 -2.959 1.00 20.10 C
+ATOM 475 CD ARG A 61 16.063 37.657 -2.503 1.00 26.00 C
+ATOM 476 NE ARG A 61 15.101 38.653 -2.063 1.00 29.28 N
+ATOM 477 CZ ARG A 61 13.794 38.351 -2.111 1.00 29.36 C
+ATOM 478 NH1 ARG A 61 13.439 37.257 -2.784 1.00 26.50 N
+ATOM 479 NH2 ARG A 61 12.925 39.249 -1.646 1.00 32.93 N
+ATOM 480 N TRP A 62 20.773 37.202 -1.273 1.00 16.51 N
+ATOM 481 CA TRP A 62 21.321 37.563 0.032 1.00 16.06 C
+ATOM 482 C TRP A 62 22.848 37.643 0.101 1.00 15.55 C
+ATOM 483 O TRP A 62 23.323 38.396 0.968 1.00 18.96 O
+ATOM 484 CB TRP A 62 20.833 36.611 1.134 1.00 17.91 C
+ATOM 485 CG TRP A 62 19.360 36.361 1.096 1.00 18.99 C
+ATOM 486 CD1 TRP A 62 18.719 35.247 0.643 1.00 20.47 C
+ATOM 487 CD2 TRP A 62 18.326 37.305 1.427 1.00 20.79 C
+ATOM 488 NE1 TRP A 62 17.360 35.457 0.609 1.00 21.70 N
+ATOM 489 CE2 TRP A 62 17.090 36.696 1.096 1.00 21.11 C
+ATOM 490 CE3 TRP A 62 18.333 38.584 1.965 1.00 19.84 C
+ATOM 491 CZ2 TRP A 62 15.875 37.327 1.307 1.00 22.57 C
+ATOM 492 CZ3 TRP A 62 17.115 39.208 2.186 1.00 23.93 C
+ATOM 493 CH2 TRP A 62 15.906 38.611 1.814 1.00 19.55 C
+ATOM 494 N TRP A 63 23.537 36.731 -0.584 1.00 16.01 N
+ATOM 495 CA TRP A 63 24.906 36.398 -0.276 1.00 15.21 C
+ATOM 496 C TRP A 63 26.089 36.924 -1.052 1.00 15.84 C
+ATOM 497 O TRP A 63 27.142 37.177 -0.432 1.00 19.13 O
+ATOM 498 CB TRP A 63 25.068 34.919 0.112 1.00 15.47 C
+ATOM 499 CG TRP A 63 24.036 34.428 1.068 1.00 12.46 C
+ATOM 500 CD1 TRP A 63 22.959 33.620 0.777 1.00 16.95 C
+ATOM 501 CD2 TRP A 63 23.919 34.728 2.450 1.00 13.27 C
+ATOM 502 NE1 TRP A 63 22.243 33.344 1.899 1.00 17.64 N
+ATOM 503 CE2 TRP A 63 22.761 34.067 2.931 1.00 17.28 C
+ATOM 504 CE3 TRP A 63 24.694 35.480 3.323 1.00 15.25 C
+ATOM 505 CZ2 TRP A 63 22.393 34.133 4.278 1.00 18.56 C
+ATOM 506 CZ3 TRP A 63 24.302 35.586 4.648 1.00 18.76 C
+ATOM 507 CH2 TRP A 63 23.179 34.910 5.117 1.00 19.36 C
+ATOM 508 N CYS A 64 25.962 36.914 -2.353 1.00 17.00 N
+ATOM 509 CA CYS A 64 26.985 37.433 -3.271 1.00 15.76 C
+ATOM 510 C CYS A 64 26.324 38.355 -4.286 1.00 15.97 C
+ATOM 511 O CYS A 64 25.102 38.314 -4.475 1.00 13.75 O
+ATOM 512 CB CYS A 64 27.638 36.265 -3.988 1.00 16.64 C
+ATOM 513 SG CYS A 64 26.562 35.233 -5.007 1.00 17.83 S
+ATOM 514 N ASN A 65 27.157 39.165 -4.908 1.00 17.73 N
+ATOM 515 CA ASN A 65 26.700 40.134 -5.920 1.00 16.46 C
+ATOM 516 C ASN A 65 26.985 39.646 -7.342 1.00 13.16 C
+ATOM 517 O ASN A 65 28.130 39.316 -7.647 1.00 15.95 O
+ATOM 518 CB ASN A 65 27.381 41.492 -5.712 1.00 19.19 C
+ATOM 519 CG ASN A 65 26.910 42.423 -6.824 1.00 19.59 C
+ATOM 520 OD1 ASN A 65 25.736 42.559 -7.141 1.00 23.26 O
+ATOM 521 ND2 ASN A 65 27.914 42.938 -7.527 1.00 25.56 N
+ATOM 522 N ASP A 66 25.920 39.484 -8.116 1.00 15.60 N
+ATOM 523 CA ASP A 66 26.102 39.180 -9.545 1.00 15.28 C
+ATOM 524 C ASP A 66 25.664 40.317 -10.460 1.00 15.23 C
+ATOM 525 O ASP A 66 25.719 40.143 -11.673 1.00 17.91 O
+ATOM 526 CB ASP A 66 25.462 37.858 -9.894 1.00 14.84 C
+ATOM 527 CG ASP A 66 23.951 37.903 -9.833 1.00 12.12 C
+ATOM 528 OD1 ASP A 66 23.288 38.898 -9.542 1.00 16.56 O
+ATOM 529 OD2 ASP A 66 23.455 36.769 -10.048 1.00 16.91 O
+ATOM 530 N GLY A 67 25.234 41.408 -9.860 1.00 18.13 N
+ATOM 531 CA GLY A 67 24.817 42.607 -10.577 1.00 19.40 C
+ATOM 532 C GLY A 67 23.544 42.553 -11.401 1.00 20.37 C
+ATOM 533 O GLY A 67 23.191 43.542 -12.055 1.00 18.23 O
+ATOM 534 N ARG A 68 22.822 41.441 -11.348 1.00 18.79 N
+ATOM 535 CA ARG A 68 21.560 41.308 -12.092 1.00 19.77 C
+ATOM 536 C ARG A 68 20.424 40.765 -11.243 1.00 18.48 C
+ATOM 537 O ARG A 68 19.385 40.383 -11.795 1.00 21.54 O
+ATOM 538 CB ARG A 68 21.746 40.454 -13.339 1.00 18.97 C
+ATOM 539 CG ARG A 68 22.197 39.048 -12.946 1.00 18.77 C
+ATOM 540 CD ARG A 68 22.477 38.187 -14.122 1.00 23.25 C
+ATOM 541 NE ARG A 68 21.439 37.201 -14.281 1.00 29.75 N
+ATOM 542 CZ ARG A 68 20.242 37.255 -14.855 1.00 28.71 C
+ATOM 543 NH1 ARG A 68 19.503 38.311 -15.145 1.00 32.34 N
+ATOM 544 NH2 ARG A 68 19.897 36.127 -15.497 1.00 33.10 N
+ATOM 545 N THR A 69 20.590 40.896 -9.952 1.00 18.68 N
+ATOM 546 CA THR A 69 19.607 40.408 -8.973 1.00 20.02 C
+ATOM 547 C THR A 69 18.994 41.597 -8.224 1.00 16.46 C
+ATOM 548 O THR A 69 19.683 42.241 -7.425 1.00 23.13 O
+ATOM 549 CB THR A 69 20.208 39.384 -7.910 1.00 19.66 C
+ATOM 550 OG1 THR A 69 20.851 38.334 -8.715 1.00 19.19 O
+ATOM 551 CG2 THR A 69 19.123 38.863 -6.955 1.00 18.58 C
+ATOM 552 N PRO A 70 17.687 41.741 -8.418 1.00 20.50 N
+ATOM 553 CA PRO A 70 16.953 42.824 -7.733 1.00 21.73 C
+ATOM 554 C PRO A 70 16.955 42.520 -6.242 1.00 22.11 C
+ATOM 555 O PRO A 70 16.739 41.356 -5.886 1.00 27.13 O
+ATOM 556 CB PRO A 70 15.557 42.749 -8.333 1.00 23.50 C
+ATOM 557 CG PRO A 70 15.759 42.092 -9.671 1.00 24.66 C
+ATOM 558 CD PRO A 70 16.887 41.095 -9.463 1.00 21.80 C
+ATOM 559 N GLY A 71 17.163 43.493 -5.401 1.00 21.67 N
+ATOM 560 CA GLY A 71 17.135 43.383 -3.947 1.00 24.03 C
+ATOM 561 C GLY A 71 18.328 42.710 -3.287 1.00 26.98 C
+ATOM 562 O GLY A 71 18.257 42.257 -2.126 1.00 32.77 O
+ATOM 563 N SER A 72 19.418 42.627 -4.018 1.00 26.40 N
+ATOM 564 CA SER A 72 20.665 41.976 -3.588 1.00 26.94 C
+ATOM 565 C SER A 72 21.139 42.582 -2.276 1.00 24.66 C
+ATOM 566 O SER A 72 21.055 43.819 -2.125 1.00 29.91 O
+ATOM 567 CB SER A 72 21.659 42.002 -4.737 1.00 25.16 C
+ATOM 568 OG SER A 72 23.015 41.835 -4.343 1.00 32.21 O
+ATOM 569 N ARG A 73 21.646 41.775 -1.369 1.00 22.51 N
+ATOM 570 CA ARG A 73 22.254 42.196 -0.113 1.00 22.60 C
+ATOM 571 C ARG A 73 23.774 42.058 0.029 1.00 22.33 C
+ATOM 572 O ARG A 73 24.425 42.749 0.849 1.00 27.92 O
+ATOM 573 CB ARG A 73 21.615 41.555 1.127 1.00 20.14 C
+ATOM 574 CG ARG A 73 20.187 41.983 1.439 1.00 22.29 C
+ATOM 575 CD ARG A 73 20.209 43.299 2.123 1.00 26.95 C
+ATOM 576 NE ARG A 73 18.928 43.771 2.617 1.00 33.74 N
+ATOM 577 CZ ARG A 73 17.980 44.302 1.823 1.00 33.76 C
+ATOM 578 NH1 ARG A 73 17.841 43.934 0.544 1.00 35.78 N
+ATOM 579 NH2 ARG A 73 17.571 45.545 2.146 1.00 33.83 N
+ATOM 580 N ASN A 74 24.346 41.143 -0.705 1.00 17.74 N
+ATOM 581 CA ASN A 74 25.780 40.833 -0.695 1.00 18.87 C
+ATOM 582 C ASN A 74 26.315 40.718 0.731 1.00 16.22 C
+ATOM 583 O ASN A 74 27.255 41.471 1.060 1.00 20.87 O
+ATOM 584 CB ASN A 74 26.565 41.786 -1.589 1.00 19.51 C
+ATOM 585 CG ASN A 74 27.982 41.328 -1.909 1.00 16.31 C
+ATOM 586 OD1 ASN A 74 28.318 40.169 -1.652 1.00 18.61 O
+ATOM 587 ND2 ASN A 74 28.838 42.192 -2.436 1.00 19.22 N
+ATOM 588 N LEU A 75 25.723 39.860 1.544 1.00 15.25 N
+ATOM 589 CA LEU A 75 26.153 39.690 2.930 1.00 14.82 C
+ATOM 590 C LEU A 75 27.518 39.011 3.085 1.00 16.78 C
+ATOM 591 O LEU A 75 28.167 39.197 4.141 1.00 22.00 O
+ATOM 592 CB LEU A 75 25.009 39.055 3.733 1.00 16.64 C
+ATOM 593 CG LEU A 75 23.815 39.998 3.979 1.00 17.08 C
+ATOM 594 CD1 LEU A 75 22.574 39.193 4.336 1.00 25.82 C
+ATOM 595 CD2 LEU A 75 24.156 40.969 5.110 1.00 20.95 C
+ATOM 596 N CYS A 76 27.973 38.312 2.061 1.00 17.66 N
+ATOM 597 CA CYS A 76 29.299 37.680 2.111 1.00 16.46 C
+ATOM 598 C CYS A 76 30.390 38.598 1.589 1.00 15.66 C
+ATOM 599 O CYS A 76 31.573 38.229 1.568 1.00 17.59 O
+ATOM 600 CB CYS A 76 29.396 36.303 1.477 1.00 17.61 C
+ATOM 601 SG CYS A 76 28.595 34.961 2.361 1.00 18.07 S
+ATOM 602 N ASN A 77 29.964 39.681 0.971 1.00 18.55 N
+ATOM 603 CA ASN A 77 30.827 40.725 0.395 1.00 19.18 C
+ATOM 604 C ASN A 77 31.711 40.163 -0.702 1.00 16.58 C
+ATOM 605 O ASN A 77 32.947 40.331 -0.689 1.00 21.61 O
+ATOM 606 CB ASN A 77 31.589 41.488 1.483 1.00 17.72 C
+ATOM 607 CG ASN A 77 32.126 42.812 0.957 1.00 20.20 C
+ATOM 608 OD1 ASN A 77 31.351 43.589 0.396 1.00 28.77 O
+ATOM 609 ND2 ASN A 77 33.430 43.012 1.047 1.00 27.05 N
+ATOM 610 N ILE A 78 31.149 39.408 -1.616 1.00 18.88 N
+ATOM 611 CA ILE A 78 31.904 38.780 -2.713 1.00 20.14 C
+ATOM 612 C ILE A 78 31.099 38.850 -4.013 1.00 17.08 C
+ATOM 613 O ILE A 78 29.864 38.688 -3.972 1.00 16.28 O
+ATOM 614 CB ILE A 78 32.183 37.255 -2.384 1.00 19.93 C
+ATOM 615 CG1 ILE A 78 30.882 36.639 -1.851 1.00 21.17 C
+ATOM 616 CG2 ILE A 78 33.437 37.003 -1.524 1.00 24.25 C
+ATOM 617 CD1 ILE A 78 30.936 35.120 -1.546 1.00 25.54 C
+ATOM 618 N PRO A 79 31.828 38.795 -5.108 1.00 16.64 N
+ATOM 619 CA PRO A 79 31.195 38.634 -6.421 1.00 19.12 C
+ATOM 620 C PRO A 79 30.792 37.146 -6.446 1.00 14.71 C
+ATOM 621 O PRO A 79 31.576 36.273 -6.005 1.00 17.48 O
+ATOM 622 CB PRO A 79 32.261 38.967 -7.445 1.00 18.74 C
+ATOM 623 CG PRO A 79 33.555 39.029 -6.710 1.00 18.97 C
+ATOM 624 CD PRO A 79 33.276 39.023 -5.215 1.00 16.79 C
+ATOM 625 N CYS A 80 29.629 36.908 -7.033 1.00 13.55 N
+ATOM 626 CA CYS A 80 29.167 35.522 -7.236 1.00 14.33 C
+ATOM 627 C CYS A 80 30.150 34.716 -8.073 1.00 13.92 C
+ATOM 628 O CYS A 80 30.214 33.484 -7.880 1.00 16.74 O
+ATOM 629 CB CYS A 80 27.749 35.338 -7.747 1.00 16.10 C
+ATOM 630 SG CYS A 80 26.471 36.160 -6.762 1.00 16.97 S
+ATOM 631 N SER A 81 30.769 35.294 -9.083 1.00 15.04 N
+ATOM 632 CA SER A 81 31.775 34.671 -9.933 1.00 15.64 C
+ATOM 633 C SER A 81 32.907 34.027 -9.109 1.00 14.90 C
+ATOM 634 O SER A 81 33.338 32.939 -9.561 1.00 21.08 O
+ATOM 635 CB SER A 81 32.381 35.643 -10.953 1.00 18.65 C
+ATOM 636 OG SER A 81 33.035 36.681 -10.235 1.00 20.86 O
+ATOM 637 N ALA A 82 33.226 34.572 -7.950 1.00 15.59 N
+ATOM 638 CA ALA A 82 34.272 33.963 -7.107 1.00 18.01 C
+ATOM 639 C ALA A 82 33.869 32.604 -6.540 1.00 18.75 C
+ATOM 640 O ALA A 82 34.703 31.798 -6.081 1.00 19.60 O
+ATOM 641 CB ALA A 82 34.722 34.930 -6.020 1.00 20.54 C
+ATOM 642 N LEU A 83 32.571 32.338 -6.519 1.00 15.36 N
+ATOM 643 CA LEU A 83 31.973 31.093 -6.042 1.00 18.66 C
+ATOM 644 C LEU A 83 32.060 29.962 -7.049 1.00 17.84 C
+ATOM 645 O LEU A 83 31.671 28.815 -6.739 1.00 21.50 O
+ATOM 646 CB LEU A 83 30.618 31.424 -5.431 1.00 17.11 C
+ATOM 647 CG LEU A 83 30.511 32.364 -4.244 1.00 16.65 C
+ATOM 648 CD1 LEU A 83 29.040 32.573 -3.857 1.00 18.78 C
+ATOM 649 CD2 LEU A 83 31.277 31.872 -3.020 1.00 20.62 C
+ATOM 650 N LEU A 84 32.473 30.271 -8.267 1.00 17.31 N
+ATOM 651 CA LEU A 84 32.610 29.295 -9.347 1.00 16.38 C
+ATOM 652 C LEU A 84 34.047 28.841 -9.570 1.00 19.37 C
+ATOM 653 O LEU A 84 34.334 28.243 -10.622 1.00 23.48 O
+ATOM 654 CB LEU A 84 31.914 29.793 -10.614 1.00 20.87 C
+ATOM 655 CG LEU A 84 30.446 30.183 -10.573 1.00 14.26 C
+ATOM 656 CD1 LEU A 84 30.014 30.771 -11.916 1.00 21.15 C
+ATOM 657 CD2 LEU A 84 29.597 28.942 -10.329 1.00 18.96 C
+ATOM 658 N SER A 85 34.942 29.197 -8.676 1.00 21.00 N
+ATOM 659 CA SER A 85 36.340 28.754 -8.727 1.00 20.86 C
+ATOM 660 C SER A 85 36.474 27.245 -8.497 1.00 19.65 C
+ATOM 661 O SER A 85 35.765 26.640 -7.681 1.00 20.62 O
+ATOM 662 CB SER A 85 37.109 29.482 -7.633 1.00 20.91 C
+ATOM 663 OG SER A 85 38.484 29.212 -7.834 1.00 26.88 O
+ATOM 664 N SER A 86 37.619 26.742 -8.943 1.00 22.57 N
+ATOM 665 CA SER A 86 38.008 25.343 -8.700 1.00 20.44 C
+ATOM 666 C SER A 86 38.388 25.153 -7.233 1.00 19.98 C
+ATOM 667 O SER A 86 38.314 24.055 -6.671 1.00 23.62 O
+ATOM 668 CB SER A 86 39.107 24.868 -9.650 1.00 23.76 C
+ATOM 669 OG SER A 86 38.401 24.215 -10.691 1.00 30.29 O
+ATOM 670 N ASP A 87 38.846 26.244 -6.668 1.00 19.74 N
+ATOM 671 CA ASP A 87 39.282 26.393 -5.271 1.00 18.72 C
+ATOM 672 C ASP A 87 37.999 26.657 -4.471 1.00 17.93 C
+ATOM 673 O ASP A 87 37.490 27.762 -4.712 1.00 20.06 O
+ATOM 674 CB ASP A 87 40.221 27.607 -5.212 1.00 21.65 C
+ATOM 675 CG ASP A 87 40.762 28.041 -3.869 1.00 23.98 C
+ATOM 676 OD1 ASP A 87 40.335 27.702 -2.742 1.00 26.26 O
+ATOM 677 OD2 ASP A 87 41.785 28.802 -3.933 1.00 32.04 O
+ATOM 678 N ILE A 88 37.732 25.850 -3.461 1.00 15.96 N
+ATOM 679 CA ILE A 88 36.515 26.089 -2.658 1.00 16.22 C
+ATOM 680 C ILE A 88 36.582 27.142 -1.563 1.00 14.43 C
+ATOM 681 O ILE A 88 35.600 27.315 -0.801 1.00 16.28 O
+ATOM 682 CB ILE A 88 35.993 24.736 -2.046 1.00 16.45 C
+ATOM 683 CG1 ILE A 88 36.920 24.200 -0.934 1.00 15.71 C
+ATOM 684 CG2 ILE A 88 35.587 23.743 -3.163 1.00 19.87 C
+ATOM 685 CD1 ILE A 88 36.363 23.226 0.137 1.00 17.47 C
+ATOM 686 N THR A 89 37.742 27.786 -1.369 1.00 14.21 N
+ATOM 687 CA THR A 89 37.913 28.772 -0.306 1.00 16.87 C
+ATOM 688 C THR A 89 36.736 29.761 -0.161 1.00 11.06 C
+ATOM 689 O THR A 89 36.341 29.983 1.000 1.00 13.83 O
+ATOM 690 CB THR A 89 39.267 29.591 -0.423 1.00 18.61 C
+ATOM 691 OG1 THR A 89 40.339 28.592 -0.514 1.00 20.24 O
+ATOM 692 CG2 THR A 89 39.419 30.553 0.766 1.00 17.79 C
+ATOM 693 N ALA A 90 36.507 30.482 -1.242 1.00 17.37 N
+ATOM 694 CA ALA A 90 35.479 31.555 -1.244 1.00 16.63 C
+ATOM 695 C ALA A 90 34.125 31.066 -0.735 1.00 14.09 C
+ATOM 696 O ALA A 90 33.366 31.671 0.065 1.00 15.68 O
+ATOM 697 CB ALA A 90 35.366 32.212 -2.617 1.00 17.13 C
+ATOM 698 N SER A 91 33.746 29.919 -1.296 1.00 13.62 N
+ATOM 699 CA SER A 91 32.494 29.220 -1.024 1.00 11.35 C
+ATOM 700 C SER A 91 32.429 28.817 0.467 1.00 11.70 C
+ATOM 701 O SER A 91 31.407 29.057 1.110 1.00 14.31 O
+ATOM 702 CB SER A 91 32.282 28.035 -1.917 1.00 13.48 C
+ATOM 703 OG SER A 91 32.020 28.335 -3.260 1.00 14.66 O
+ATOM 704 N VAL A 92 33.502 28.211 0.953 1.00 13.17 N
+ATOM 705 CA VAL A 92 33.595 27.846 2.381 1.00 13.44 C
+ATOM 706 C VAL A 92 33.477 29.105 3.261 1.00 12.00 C
+ATOM 707 O VAL A 92 32.716 29.020 4.244 1.00 13.83 O
+ATOM 708 CB VAL A 92 34.890 27.038 2.650 1.00 11.69 C
+ATOM 709 CG1 VAL A 92 35.125 26.987 4.151 1.00 16.05 C
+ATOM 710 CG2 VAL A 92 34.776 25.672 2.013 1.00 15.64 C
+ATOM 711 N ASN A 93 34.199 30.163 2.912 1.00 15.12 N
+ATOM 712 CA ASN A 93 34.160 31.413 3.685 1.00 15.10 C
+ATOM 713 C ASN A 93 32.747 32.034 3.803 1.00 9.80 C
+ATOM 714 O ASN A 93 32.361 32.393 4.908 1.00 16.51 O
+ATOM 715 CB ASN A 93 35.168 32.472 3.247 1.00 16.33 C
+ATOM 716 CG ASN A 93 36.582 32.083 3.642 1.00 19.02 C
+ATOM 717 OD1 ASN A 93 37.546 32.671 3.120 1.00 29.57 O
+ATOM 718 ND2 ASN A 93 36.710 31.188 4.631 1.00 24.29 N
+ATOM 719 N CYS A 94 32.096 31.996 2.670 1.00 15.72 N
+ATOM 720 CA CYS A 94 30.695 32.449 2.633 1.00 13.58 C
+ATOM 721 C CYS A 94 29.778 31.527 3.424 1.00 13.05 C
+ATOM 722 O CYS A 94 28.973 32.025 4.236 1.00 16.42 O
+ATOM 723 CB CYS A 94 30.291 32.704 1.185 1.00 12.05 C
+ATOM 724 SG CYS A 94 28.644 33.431 1.046 1.00 15.81 S
+ATOM 725 N ALA A 95 29.945 30.225 3.263 1.00 13.56 N
+ATOM 726 CA ALA A 95 29.134 29.227 3.969 1.00 15.63 C
+ATOM 727 C ALA A 95 29.236 29.330 5.491 1.00 10.80 C
+ATOM 728 O ALA A 95 28.222 29.180 6.179 1.00 12.93 O
+ATOM 729 CB ALA A 95 29.495 27.794 3.612 1.00 14.09 C
+ATOM 730 N LYS A 96 30.424 29.628 5.987 1.00 11.19 N
+ATOM 731 CA LYS A 96 30.627 29.912 7.419 1.00 13.27 C
+ATOM 732 C LYS A 96 29.802 31.115 7.906 1.00 13.38 C
+ATOM 733 O LYS A 96 29.296 31.011 9.027 1.00 15.75 O
+ATOM 734 CB LYS A 96 32.100 30.112 7.775 1.00 12.67 C
+ATOM 735 CG LYS A 96 32.874 28.792 7.697 1.00 11.93 C
+ATOM 736 CD LYS A 96 34.358 29.071 7.879 1.00 15.55 C
+ATOM 737 CE LYS A 96 35.205 27.816 7.938 1.00 18.03 C
+ATOM 738 NZ LYS A 96 36.610 28.242 8.169 1.00 20.72 N
+ATOM 739 N LYS A 97 29.660 32.101 7.049 1.00 12.90 N
+ATOM 740 CA LYS A 97 28.811 33.263 7.379 1.00 16.43 C
+ATOM 741 C LYS A 97 27.333 32.905 7.386 1.00 18.88 C
+ATOM 742 O LYS A 97 26.570 33.318 8.272 1.00 19.20 O
+ATOM 743 CB LYS A 97 29.087 34.456 6.470 1.00 18.97 C
+ATOM 744 CG LYS A 97 30.537 34.927 6.563 1.00 19.81 C
+ATOM 745 CD LYS A 97 30.841 36.073 5.610 1.00 22.00 C
+ATOM 746 CE LYS A 97 32.340 36.293 5.510 1.00 23.71 C
+ATOM 747 NZ LYS A 97 32.569 37.524 4.708 1.00 29.75 N
+ATOM 748 N ILE A 98 26.897 32.127 6.416 1.00 15.72 N
+ATOM 749 CA ILE A 98 25.520 31.693 6.266 1.00 16.36 C
+ATOM 750 C ILE A 98 25.030 30.871 7.453 1.00 16.70 C
+ATOM 751 O ILE A 98 24.059 31.254 8.126 1.00 18.80 O
+ATOM 752 CB ILE A 98 25.307 30.997 4.896 1.00 15.14 C
+ATOM 753 CG1 ILE A 98 25.643 31.910 3.702 1.00 13.63 C
+ATOM 754 CG2 ILE A 98 23.887 30.386 4.817 1.00 15.62 C
+ATOM 755 CD1 ILE A 98 25.620 31.204 2.321 1.00 17.82 C
+ATOM 756 N VAL A 99 25.875 29.966 7.915 1.00 16.73 N
+ATOM 757 CA VAL A 99 25.488 29.009 8.955 1.00 18.71 C
+ATOM 758 C VAL A 99 25.431 29.697 10.316 1.00 20.36 C
+ATOM 759 O VAL A 99 24.737 29.212 11.233 1.00 25.30 O
+ATOM 760 CB VAL A 99 26.398 27.774 8.859 1.00 16.94 C
+ATOM 761 CG1 VAL A 99 27.811 28.138 9.279 1.00 19.19 C
+ATOM 762 CG2 VAL A 99 25.834 26.594 9.632 1.00 18.97 C
+ATOM 763 N SER A 100 26.205 30.752 10.409 1.00 17.04 N
+ATOM 764 CA SER A 100 26.297 31.529 11.650 1.00 23.81 C
+ATOM 765 C SER A 100 25.124 32.495 11.765 1.00 24.03 C
+ATOM 766 O SER A 100 24.995 33.131 12.820 1.00 28.66 O
+ATOM 767 CB SER A 100 27.647 32.194 11.723 1.00 19.71 C
+ATOM 768 OG SER A 100 28.714 31.264 11.818 1.00 25.04 O
+ATOM 769 N ASP A 101 24.307 32.599 10.750 1.00 25.78 N
+ATOM 770 CA ASP A 101 23.162 33.495 10.650 1.00 26.98 C
+ATOM 771 C ASP A 101 21.924 33.183 11.481 1.00 28.34 C
+ATOM 772 O ASP A 101 21.132 34.143 11.678 1.00 31.88 O
+ATOM 773 CB ASP A 101 22.806 33.854 9.207 1.00 27.75 C
+ATOM 774 CG ASP A 101 22.426 35.320 9.009 1.00 30.86 C
+ATOM 775 OD1 ASP A 101 23.248 36.223 9.266 1.00 34.67 O
+ATOM 776 OD2 ASP A 101 21.276 35.519 8.551 1.00 32.70 O
+ATOM 777 N GLY A 102 21.723 31.942 11.887 1.00 28.87 N
+ATOM 778 CA GLY A 102 20.622 31.656 12.823 1.00 30.67 C
+ATOM 779 C GLY A 102 19.812 30.413 12.505 1.00 27.92 C
+ATOM 780 O GLY A 102 19.195 29.898 13.458 1.00 30.42 O
+ATOM 781 N ASN A 103 19.805 30.005 11.244 1.00 29.26 N
+ATOM 782 CA ASN A 103 19.076 28.799 10.848 1.00 25.22 C
+ATOM 783 C ASN A 103 19.990 27.600 10.597 1.00 22.06 C
+ATOM 784 O ASN A 103 19.472 26.579 10.117 1.00 24.23 O
+ATOM 785 CB ASN A 103 17.976 29.046 9.824 1.00 24.87 C
+ATOM 786 CG ASN A 103 16.708 28.245 10.063 1.00 27.08 C
+ATOM 787 OD1 ASN A 103 16.513 27.577 11.098 1.00 30.30 O
+ATOM 788 ND2 ASN A 103 15.672 28.428 9.240 1.00 30.15 N
+ATOM 789 N GLY A 104 21.240 27.711 10.997 1.00 23.20 N
+ATOM 790 CA GLY A 104 22.177 26.567 10.860 1.00 19.51 C
+ATOM 791 C GLY A 104 22.173 26.142 9.388 1.00 15.58 C
+ATOM 792 O GLY A 104 22.151 27.040 8.541 1.00 22.50 O
+ATOM 793 N MET A 105 22.185 24.847 9.115 1.00 17.51 N
+ATOM 794 CA MET A 105 22.278 24.370 7.724 1.00 16.07 C
+ATOM 795 C MET A 105 20.915 24.309 7.063 1.00 13.35 C
+ATOM 796 O MET A 105 20.902 23.921 5.871 1.00 15.50 O
+ATOM 797 CB MET A 105 23.036 23.048 7.632 1.00 19.32 C
+ATOM 798 CG MET A 105 24.519 23.221 7.871 1.00 17.10 C
+ATOM 799 SD MET A 105 25.379 21.660 7.491 1.00 20.27 S
+ATOM 800 CE MET A 105 25.369 21.706 5.703 1.00 20.03 C
+ATOM 801 N ASN A 106 19.896 24.839 7.735 1.00 14.64 N
+ATOM 802 CA ASN A 106 18.557 24.898 7.103 1.00 18.58 C
+ATOM 803 C ASN A 106 18.566 25.831 5.892 1.00 18.20 C
+ATOM 804 O ASN A 106 17.678 25.748 5.021 1.00 22.14 O
+ATOM 805 CB ASN A 106 17.420 25.150 8.084 1.00 19.18 C
+ATOM 806 CG ASN A 106 17.207 23.938 8.967 1.00 19.21 C
+ATOM 807 OD1 ASN A 106 16.836 22.867 8.443 1.00 24.65 O
+ATOM 808 ND2 ASN A 106 17.542 24.047 10.255 1.00 25.28 N
+ATOM 809 N ALA A 107 19.617 26.647 5.800 1.00 18.54 N
+ATOM 810 CA ALA A 107 19.824 27.514 4.634 1.00 18.74 C
+ATOM 811 C ALA A 107 20.007 26.744 3.329 1.00 16.80 C
+ATOM 812 O ALA A 107 19.734 27.292 2.260 1.00 20.63 O
+ATOM 813 CB ALA A 107 21.060 28.374 4.877 1.00 19.47 C
+ATOM 814 N TRP A 108 20.371 25.465 3.434 1.00 16.46 N
+ATOM 815 CA TRP A 108 20.532 24.624 2.241 1.00 14.68 C
+ATOM 816 C TRP A 108 19.317 23.688 2.110 1.00 17.55 C
+ATOM 817 O TRP A 108 19.240 22.816 2.993 1.00 20.05 O
+ATOM 818 CB TRP A 108 21.840 23.826 2.338 1.00 16.18 C
+ATOM 819 CG TRP A 108 23.000 24.736 2.026 1.00 15.50 C
+ATOM 820 CD1 TRP A 108 23.360 25.234 0.803 1.00 15.45 C
+ATOM 821 CD2 TRP A 108 23.798 25.437 2.991 1.00 15.57 C
+ATOM 822 NE1 TRP A 108 24.414 26.102 0.952 1.00 17.40 N
+ATOM 823 CE2 TRP A 108 24.711 26.237 2.272 1.00 15.78 C
+ATOM 824 CE3 TRP A 108 23.833 25.426 4.379 1.00 12.79 C
+ATOM 825 CZ2 TRP A 108 25.682 26.985 2.917 1.00 15.77 C
+ATOM 826 CZ3 TRP A 108 24.773 26.200 5.033 1.00 16.28 C
+ATOM 827 CH2 TRP A 108 25.691 26.960 4.298 1.00 16.65 C
+ATOM 828 N VAL A 109 18.487 23.925 1.101 1.00 15.35 N
+ATOM 829 CA VAL A 109 17.265 23.096 1.025 1.00 19.96 C
+ATOM 830 C VAL A 109 17.573 21.611 0.884 1.00 17.16 C
+ATOM 831 O VAL A 109 16.923 20.811 1.587 1.00 20.77 O
+ATOM 832 CB VAL A 109 16.146 23.660 0.145 1.00 21.94 C
+ATOM 833 CG1 VAL A 109 16.607 23.905 -1.285 1.00 27.84 C
+ATOM 834 CG2 VAL A 109 14.901 22.783 0.136 1.00 21.22 C
+ATOM 835 N ALA A 110 18.562 21.308 0.075 1.00 20.97 N
+ATOM 836 CA ALA A 110 18.972 19.935 -0.237 1.00 18.78 C
+ATOM 837 C ALA A 110 19.502 19.244 1.006 1.00 18.30 C
+ATOM 838 O ALA A 110 19.197 18.046 1.174 1.00 18.47 O
+ATOM 839 CB ALA A 110 19.857 19.777 -1.455 1.00 20.83 C
+ATOM 840 N TRP A 111 20.121 19.983 1.892 1.00 16.62 N
+ATOM 841 CA TRP A 111 20.551 19.500 3.202 1.00 15.74 C
+ATOM 842 C TRP A 111 19.343 19.085 4.046 1.00 19.12 C
+ATOM 843 O TRP A 111 19.284 17.986 4.643 1.00 16.20 O
+ATOM 844 CB TRP A 111 21.486 20.451 3.925 1.00 14.63 C
+ATOM 845 CG TRP A 111 21.858 19.904 5.252 1.00 14.87 C
+ATOM 846 CD1 TRP A 111 22.856 18.975 5.486 1.00 18.11 C
+ATOM 847 CD2 TRP A 111 21.199 20.093 6.504 1.00 16.63 C
+ATOM 848 NE1 TRP A 111 22.848 18.592 6.808 1.00 17.16 N
+ATOM 849 CE2 TRP A 111 21.798 19.221 7.435 1.00 16.77 C
+ATOM 850 CE3 TRP A 111 20.182 20.959 6.908 1.00 15.16 C
+ATOM 851 CZ2 TRP A 111 21.492 19.284 8.784 1.00 17.69 C
+ATOM 852 CZ3 TRP A 111 19.818 20.954 8.240 1.00 16.88 C
+ATOM 853 CH2 TRP A 111 20.443 20.110 9.162 1.00 19.73 C
+ATOM 854 N ARG A 112 18.438 20.038 4.228 1.00 17.47 N
+ATOM 855 CA ARG A 112 17.185 19.830 4.955 1.00 19.60 C
+ATOM 856 C ARG A 112 16.472 18.593 4.391 1.00 17.68 C
+ATOM 857 O ARG A 112 16.000 17.792 5.220 1.00 22.77 O
+ATOM 858 CB ARG A 112 16.214 20.990 4.944 1.00 17.57 C
+ATOM 859 CG ARG A 112 16.592 22.438 5.091 1.00 25.27 C
+ATOM 860 CD ARG A 112 15.368 23.296 5.135 1.00 21.23 C
+ATOM 861 NE ARG A 112 14.776 23.555 3.836 1.00 28.06 N
+ATOM 862 CZ ARG A 112 14.785 24.695 3.143 1.00 27.89 C
+ATOM 863 NH1 ARG A 112 15.596 25.721 3.383 1.00 30.12 N
+ATOM 864 NH2 ARG A 112 13.717 25.008 2.398 1.00 30.39 N
+ATOM 865 N ASN A 113 16.307 18.521 3.074 1.00 19.43 N
+ATOM 866 CA ASN A 113 15.464 17.463 2.491 1.00 20.16 C
+ATOM 867 C ASN A 113 16.111 16.083 2.586 1.00 21.26 C
+ATOM 868 O ASN A 113 15.454 15.054 2.808 1.00 24.90 O
+ATOM 869 CB ASN A 113 14.966 17.779 1.085 1.00 20.43 C
+ATOM 870 CG ASN A 113 14.003 18.961 1.018 1.00 16.63 C
+ATOM 871 OD1 ASN A 113 13.454 19.344 2.059 1.00 25.68 O
+ATOM 872 ND2 ASN A 113 13.840 19.555 -0.159 1.00 22.00 N
+ATOM 873 N ARG A 114 17.401 16.041 2.355 1.00 19.72 N
+ATOM 874 CA ARG A 114 18.156 14.836 2.042 1.00 21.43 C
+ATOM 875 C ARG A 114 19.258 14.440 2.991 1.00 22.71 C
+ATOM 876 O ARG A 114 19.505 13.217 3.097 1.00 24.85 O
+ATOM 877 CB ARG A 114 18.622 14.910 0.576 1.00 21.05 C
+ATOM 878 CG ARG A 114 17.395 14.614 -0.300 1.00 26.59 C
+ATOM 879 CD ARG A 114 17.729 14.399 -1.731 1.00 26.92 C
+ATOM 880 NE ARG A 114 18.153 15.677 -2.301 1.00 33.46 N
+ATOM 881 CZ ARG A 114 17.826 16.080 -3.535 1.00 32.00 C
+ATOM 882 NH1 ARG A 114 17.378 15.225 -4.456 1.00 36.02 N
+ATOM 883 NH2 ARG A 114 17.735 17.388 -3.796 1.00 36.05 N
+ATOM 884 N CYS A 115 19.743 15.372 3.773 1.00 17.56 N
+ATOM 885 CA CYS A 115 20.843 15.041 4.708 1.00 15.08 C
+ATOM 886 C CYS A 115 20.448 14.933 6.159 1.00 16.64 C
+ATOM 887 O CYS A 115 20.972 14.116 6.940 1.00 21.71 O
+ATOM 888 CB CYS A 115 21.991 16.018 4.426 1.00 15.21 C
+ATOM 889 SG CYS A 115 22.563 16.009 2.739 1.00 19.35 S
+ATOM 890 N LYS A 116 19.714 15.918 6.619 1.00 18.76 N
+ATOM 891 CA LYS A 116 19.332 16.085 8.025 1.00 19.14 C
+ATOM 892 C LYS A 116 18.634 14.821 8.518 1.00 21.91 C
+ATOM 893 O LYS A 116 17.819 14.234 7.785 1.00 24.78 O
+ATOM 894 CB LYS A 116 18.492 17.363 8.126 1.00 21.01 C
+ATOM 895 CG LYS A 116 17.930 17.512 9.547 1.00 21.28 C
+ATOM 896 CD LYS A 116 16.745 18.481 9.554 1.00 25.86 C
+ATOM 897 CE LYS A 116 16.658 19.147 10.918 1.00 25.58 C
+ATOM 898 NZ LYS A 116 15.454 20.010 11.047 1.00 34.69 N
+ATOM 899 N GLY A 117 19.152 14.318 9.635 1.00 26.39 N
+ATOM 900 CA GLY A 117 18.558 13.126 10.267 1.00 29.06 C
+ATOM 901 C GLY A 117 19.018 11.781 9.733 1.00 28.29 C
+ATOM 902 O GLY A 117 18.499 10.733 10.164 1.00 31.90 O
+ATOM 903 N THR A 118 19.892 11.802 8.740 1.00 26.88 N
+ATOM 904 CA THR A 118 20.473 10.578 8.171 1.00 22.31 C
+ATOM 905 C THR A 118 21.868 10.375 8.761 1.00 21.43 C
+ATOM 906 O THR A 118 22.321 11.119 9.650 1.00 22.31 O
+ATOM 907 CB THR A 118 20.440 10.571 6.598 1.00 18.59 C
+ATOM 908 OG1 THR A 118 21.560 11.404 6.161 1.00 22.71 O
+ATOM 909 CG2 THR A 118 19.095 11.104 6.074 1.00 21.13 C
+ATOM 910 N ASP A 119 22.392 9.213 8.431 1.00 21.20 N
+ATOM 911 CA ASP A 119 23.768 8.830 8.756 1.00 22.61 C
+ATOM 912 C ASP A 119 24.713 9.543 7.779 1.00 20.13 C
+ATOM 913 O ASP A 119 25.178 8.950 6.780 1.00 20.57 O
+ATOM 914 CB ASP A 119 23.934 7.313 8.738 1.00 21.00 C
+ATOM 915 CG ASP A 119 25.347 6.900 9.121 1.00 25.32 C
+ATOM 916 OD1 ASP A 119 26.051 7.633 9.830 1.00 27.93 O
+ATOM 917 OD2 ASP A 119 25.715 5.746 8.804 1.00 27.31 O
+ATOM 918 N VAL A 120 24.988 10.793 8.094 1.00 23.41 N
+ATOM 919 CA VAL A 120 25.886 11.637 7.306 1.00 20.10 C
+ATOM 920 C VAL A 120 27.342 11.199 7.304 1.00 18.51 C
+ATOM 921 O VAL A 120 28.099 11.604 6.406 1.00 19.95 O
+ATOM 922 CB VAL A 120 25.721 13.134 7.630 1.00 19.91 C
+ATOM 923 CG1 VAL A 120 24.356 13.655 7.183 1.00 23.59 C
+ATOM 924 CG2 VAL A 120 26.088 13.478 9.055 1.00 20.79 C
+ATOM 925 N GLN A 121 27.701 10.430 8.306 1.00 21.83 N
+ATOM 926 CA GLN A 121 29.021 9.783 8.389 1.00 21.12 C
+ATOM 927 C GLN A 121 29.317 8.861 7.207 1.00 20.78 C
+ATOM 928 O GLN A 121 30.480 8.669 6.820 1.00 19.66 O
+ATOM 929 CB GLN A 121 29.088 9.048 9.728 1.00 24.21 C
+ATOM 930 CG GLN A 121 30.530 8.965 10.167 1.00 26.13 C
+ATOM 931 CD GLN A 121 30.615 8.877 11.674 1.00 26.94 C
+ATOM 932 OE1 GLN A 121 31.368 9.632 12.283 1.00 31.43 O
+ATOM 933 NE2 GLN A 121 29.884 7.871 12.151 1.00 28.26 N
+ATOM 934 N ALA A 122 28.300 8.271 6.576 1.00 17.62 N
+ATOM 935 CA ALA A 122 28.390 7.548 5.311 1.00 19.46 C
+ATOM 936 C ALA A 122 29.186 8.290 4.227 1.00 17.98 C
+ATOM 937 O ALA A 122 30.031 7.710 3.523 1.00 21.35 O
+ATOM 938 CB ALA A 122 26.999 7.244 4.783 1.00 18.29 C
+ATOM 939 N TRP A 123 29.021 9.616 4.240 1.00 18.07 N
+ATOM 940 CA TRP A 123 29.703 10.502 3.283 1.00 16.76 C
+ATOM 941 C TRP A 123 31.218 10.581 3.392 1.00 16.76 C
+ATOM 942 O TRP A 123 31.905 10.942 2.412 1.00 20.08 O
+ATOM 943 CB TRP A 123 29.027 11.872 3.347 1.00 19.49 C
+ATOM 944 CG TRP A 123 27.621 11.735 2.850 1.00 17.43 C
+ATOM 945 CD1 TRP A 123 26.485 11.608 3.588 1.00 19.42 C
+ATOM 946 CD2 TRP A 123 27.241 11.547 1.481 1.00 17.47 C
+ATOM 947 NE1 TRP A 123 25.405 11.458 2.774 1.00 18.86 N
+ATOM 948 CE2 TRP A 123 25.827 11.391 1.479 1.00 17.28 C
+ATOM 949 CE3 TRP A 123 27.947 11.468 0.283 1.00 20.03 C
+ATOM 950 CZ2 TRP A 123 25.111 11.184 0.311 1.00 18.33 C
+ATOM 951 CZ3 TRP A 123 27.222 11.329 -0.886 1.00 20.85 C
+ATOM 952 CH2 TRP A 123 25.835 11.127 -0.869 1.00 20.45 C
+ATOM 953 N ILE A 124 31.741 10.269 4.554 1.00 14.19 N
+ATOM 954 CA ILE A 124 33.186 10.292 4.801 1.00 16.82 C
+ATOM 955 C ILE A 124 33.863 8.955 5.024 1.00 18.45 C
+ATOM 956 O ILE A 124 35.100 8.892 5.134 1.00 21.02 O
+ATOM 957 CB ILE A 124 33.504 11.403 5.863 1.00 17.69 C
+ATOM 958 CG1 ILE A 124 32.956 10.984 7.234 1.00 18.86 C
+ATOM 959 CG2 ILE A 124 33.024 12.804 5.387 1.00 21.01 C
+ATOM 960 CD1 ILE A 124 33.729 11.437 8.488 1.00 22.81 C
+ATOM 961 N ARG A 125 33.080 7.898 5.131 1.00 21.05 N
+ATOM 962 CA ARG A 125 33.594 6.534 5.320 1.00 19.12 C
+ATOM 963 C ARG A 125 34.311 6.113 4.036 1.00 17.52 C
+ATOM 964 O ARG A 125 33.843 6.337 2.906 1.00 23.44 O
+ATOM 965 CB ARG A 125 32.476 5.551 5.650 1.00 19.54 C
+ATOM 966 CG ARG A 125 32.117 5.596 7.145 1.00 22.40 C
+ATOM 967 CD ARG A 125 31.277 4.392 7.482 1.00 24.05 C
+ATOM 968 NE ARG A 125 30.282 4.754 8.466 1.00 28.08 N
+ATOM 969 CZ ARG A 125 28.984 4.993 8.331 1.00 23.40 C
+ATOM 970 NH1 ARG A 125 28.334 4.690 7.207 1.00 23.69 N
+ATOM 971 NH2 ARG A 125 28.392 5.549 9.392 1.00 23.77 N
+ATOM 972 N GLY A 126 35.497 5.568 4.243 1.00 18.80 N
+ATOM 973 CA GLY A 126 36.291 5.058 3.102 1.00 21.28 C
+ATOM 974 C GLY A 126 37.334 6.066 2.658 1.00 22.50 C
+ATOM 975 O GLY A 126 38.220 5.729 1.855 1.00 23.59 O
+ATOM 976 N CYS A 127 37.335 7.221 3.297 1.00 19.05 N
+ATOM 977 CA CYS A 127 38.234 8.333 2.961 1.00 19.13 C
+ATOM 978 C CYS A 127 39.422 8.382 3.925 1.00 22.50 C
+ATOM 979 O CYS A 127 39.206 8.267 5.138 1.00 21.64 O
+ATOM 980 CB CYS A 127 37.453 9.628 2.990 1.00 18.75 C
+ATOM 981 SG CYS A 127 36.010 9.816 1.936 1.00 19.93 S
+ATOM 982 N ARG A 128 40.586 8.695 3.393 1.00 23.60 N
+ATOM 983 CA ARG A 128 41.774 8.960 4.217 1.00 28.29 C
+ATOM 984 C ARG A 128 41.820 10.438 4.578 1.00 25.64 C
+ATOM 985 O ARG A 128 41.976 11.291 3.694 1.00 30.98 O
+ATOM 986 CB ARG A 128 43.047 8.304 3.707 1.00 30.82 C
+ATOM 987 CG ARG A 128 43.231 6.886 4.280 1.00 34.25 C
+ATOM 988 CD ARG A 128 43.833 6.911 5.651 1.00 33.59 C
+ATOM 989 NE ARG A 128 45.246 7.263 5.636 1.00 37.63 N
+ATOM 990 CZ ARG A 128 45.862 8.258 6.281 1.00 38.37 C
+ATOM 991 NH1 ARG A 128 45.241 9.069 7.151 1.00 38.97 N
+ATOM 992 NH2 ARG A 128 47.134 8.554 5.973 1.00 40.22 N
+ATOM 993 N LEU A 129 41.289 10.715 5.771 1.00 26.05 N
+ATOM 994 CA LEU A 129 41.094 12.084 6.273 1.00 26.89 C
+ATOM 995 C LEU A 129 42.119 12.382 7.370 1.00 29.58 C
+ATOM 996 O LEU A 129 41.730 12.276 8.559 1.00 33.54 O
+ATOM 997 CB LEU A 129 39.635 12.335 6.646 1.00 26.31 C
+ATOM 998 CG LEU A 129 38.689 12.917 5.620 1.00 23.49 C
+ATOM 999 CD1 LEU A 129 39.112 12.657 4.191 1.00 26.43 C
+ATOM 1000 CD2 LEU A 129 37.310 12.325 5.886 1.00 25.15 C
+ATOM 1001 OXT LEU A 129 43.232 12.675 6.905 1.00 34.20 O
+TER 1002 LEU A 129
+HETATM 1003 O HOH A 130 23.434 40.063 -6.661 1.00 19.48 O
+HETATM 1004 O HOH A 131 31.994 26.416 -6.047 0.90 22.43 O
+HETATM 1005 O HOH A 132 30.250 13.337 9.787 0.98 20.93 O
+HETATM 1006 O HOH A 133 22.384 42.331 -8.165 0.90 21.85 O
+HETATM 1007 O HOH A 134 29.239 27.621 -3.670 1.00 17.47 O
+HETATM 1008 O HOH A 135 29.464 37.761 -10.492 0.98 20.05 O
+HETATM 1009 O HOH A 136 20.807 36.305 -11.082 1.00 18.47 O
+HETATM 1010 O HOH A 137 41.318 17.849 -1.378 0.98 20.99 O
+HETATM 1011 O HOH A 138 34.697 29.056 -4.039 0.89 22.31 O
+HETATM 1012 O HOH A 139 26.871 17.298 13.496 1.00 20.31 O
+HETATM 1013 O HOH A 140 32.131 11.050 -5.817 0.97 21.39 O
+HETATM 1014 O HOH A 141 23.468 40.040 -2.372 0.91 23.40 O
+HETATM 1015 O HOH A 142 21.390 45.524 -11.035 0.96 20.63 O
+HETATM 1016 O HOH A 143 34.490 26.578 -5.741 0.75 22.11 O
+HETATM 1017 O HOH A 144 16.422 34.139 -3.527 0.91 20.71 O
+HETATM 1018 O HOH A 145 21.374 29.926 8.946 0.83 24.21 O
+HETATM 1019 O HOH A 146 41.048 12.539 -0.011 0.70 22.71 O
+HETATM 1020 O HOH A 147 32.794 35.686 2.558 0.78 20.71 O
+HETATM 1021 O HOH A 148 49.648 8.964 6.343 0.83 21.93 O
+HETATM 1022 O HOH A 149 14.452 34.901 -13.339 0.69 23.89 O
+HETATM 1023 O HOH A 150 22.930 10.839 4.044 0.92 22.02 O
+HETATM 1024 O HOH A 151 16.012 18.490 -2.200 0.85 24.37 O
+HETATM 1025 O HOH A 152 12.130 21.587 3.044 0.78 24.35 O
+HETATM 1026 O HOH A 153 15.684 38.922 -5.813 0.76 24.50 O
+HETATM 1027 O HOH A 154 10.652 24.228 3.428 0.80 21.12 O
+HETATM 1028 O HOH A 155 44.070 17.975 2.852 0.80 21.64 O
+HETATM 1029 O HOH A 156 32.029 13.080 -8.110 0.85 20.63 O
+HETATM 1030 O HOH A 157 36.425 19.613 15.174 0.56 23.44 O
+HETATM 1031 O HOH A 158 37.941 30.505 -3.686 0.79 21.54 O
+HETATM 1032 O HOH A 159 30.710 42.741 -6.289 0.72 22.79 O
+HETATM 1033 O HOH A 160 23.922 44.367 -7.653 0.62 22.78 O
+HETATM 1034 O HOH A 161 33.829 34.252 0.626 0.73 20.81 O
+HETATM 1035 O HOH A 162 29.613 40.730 -9.602 0.78 22.12 O
+HETATM 1036 O HOH A 163 23.563 7.995 4.406 0.58 22.93 O
+HETATM 1037 O HOH A 164 31.511 42.362 -4.183 0.73 22.01 O
+HETATM 1038 O HOH A 165 21.882 29.536 -15.013 0.81 22.04 O
+HETATM 1039 O HOH A 166 37.763 20.913 9.782 0.86 21.57 O
+HETATM 1040 O HOH A 167 42.338 17.481 5.165 0.65 22.17 O
+HETATM 1041 O HOH A 168 23.344 39.739 -4.358 0.72 21.56 O
+HETATM 1042 O HOH A 169 22.984 29.224 13.124 0.75 22.56 O
+HETATM 1043 O HOH A 170 30.778 7.794 -3.514 0.65 21.58 O
+HETATM 1044 O HOH A 171 42.965 14.657 4.991 0.63 23.91 O
+HETATM 1045 O HOH A 172 36.927 17.948 -13.093 0.62 23.36 O
+HETATM 1046 O HOH A 173 35.412 25.852 -11.575 0.58 23.42 O
+HETATM 1047 O HOH A 174 37.428 32.540 -5.787 0.62 21.98 O
+HETATM 1048 O HOH A 175 37.317 8.592 7.456 0.64 22.92 O
+HETATM 1049 O HOH A 176 9.314 36.705 -11.546 0.69 23.77 O
+HETATM 1050 O HOH A 177 39.972 23.760 -2.655 0.86 18.96 O
+HETATM 1051 O HOH A 178 22.128 30.274 -0.543 0.76 18.78 O
+HETATM 1052 O HOH A 179 22.244 15.813 10.000 0.68 19.66 O
+HETATM 1053 O HOH A 180 40.729 9.223 0.292 0.64 20.15 O
+HETATM 1054 O HOH A 181 12.500 15.267 4.097 0.56 20.12 O
+HETATM 1055 O HOH A 182 20.372 28.618 -2.353 0.64 20.17 O
+HETATM 1056 O HOH A 183 22.793 15.462 -6.673 0.63 20.60 O
+HETATM 1057 O HOH A 184 23.138 31.809 15.121 0.55 20.90 O
+HETATM 1058 O HOH A 185 22.671 38.691 8.245 0.48 21.16 O
+HETATM 1059 O HOH A 186 33.966 33.112 6.837 0.59 19.45 O
+HETATM 1060 O HOH A 187 19.572 25.423 -1.420 0.53 19.94 O
+HETATM 1061 O HOH A 188 14.790 15.672 7.259 0.52 21.22 O
+HETATM 1062 O HOH A 189 19.112 28.022 -14.647 0.49 19.83 O
+HETATM 1063 O HOH A 190 17.302 39.059 -12.453 0.52 20.14 O
+HETATM 1064 O HOH A 191 16.198 14.502 5.577 0.46 20.78 O
+HETATM 1065 O HOH A 192 17.345 46.346 -7.080 0.50 18.13 O
+HETATM 1066 O HOH A 193 14.992 31.300 -4.242 0.46 17.90 O
+HETATM 1067 O HOH A 194 28.196 44.775 -3.148 0.44 18.15 O
+HETATM 1068 O HOH A 195 29.479 13.863 -9.107 0.44 18.30 O
+HETATM 1069 O HOH A 196 23.613 44.811 2.608 0.45 17.66 O
+HETATM 1070 O HOH A 197 40.572 22.184 -6.358 0.42 18.06 O
+HETATM 1071 O HOH A 198 12.475 31.860 -6.226 0.47 17.85 O
+HETATM 1072 O HOH A 199 16.684 13.594 -5.832 0.31 18.51 O
+HETATM 1073 O HOH A 200 27.534 38.059 -12.862 0.48 18.19 O
+HETATM 1074 O HOH A 201 25.892 35.973 11.563 0.46 18.15 O
+HETATM 1075 O HOH A 202 24.790 25.182 16.063 0.46 17.64 O
+HETATM 1076 O HOH A 203 12.580 21.214 5.006 0.51 17.97 O
+HETATM 1077 O HOH A 204 19.687 23.750 -4.851 0.37 18.08 O
+HETATM 1078 O HOH A 205 27.098 35.956 -12.358 0.39 18.71 O
+HETATM 1079 O HOH A 206 37.255 9.634 10.002 0.46 18.39 O
+HETATM 1080 O HOH A 207 43.755 23.843 8.038 0.38 17.96 O
+CONECT 48 981
+CONECT 238 889
+CONECT 513 630
+CONECT 601 724
+CONECT 630 513
+CONECT 724 601
+CONECT 889 238
+CONECT 981 48
+MASTER 290 0 0 8 2 0 0 6 1079 1 8 10
+END
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/ions.mdp b/containers/2_ApplicationSpecific/GROMACS/inputs/ions.mdp
new file mode 100644
index 00000000..eb2cfc5e
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/ions.mdp
@@ -0,0 +1,15 @@
+; ions.mdp - used as input into grompp to generate ions.tpr
+; Parameters describing what to do, when to stop and what to save
+integrator = steep ; Algorithm (steep = steepest descent minimization)
+emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
+emstep = 0.01 ; Minimization step size
+nsteps = 50000 ; Maximum number of (minimization) steps to perform
+
+; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
+nstlist = 1 ; Frequency to update the neighbor list and long range forces
+cutoff-scheme = Verlet ; Buffered neighbor searching
+ns_type = grid ; Method to determine neighbor list (simple, grid)
+coulombtype = cutoff ; Treatment of long range electrostatic interactions
+rcoulomb = 1.0 ; Short-range electrostatic cut-off
+rvdw = 1.0 ; Short-range Van der Waals cut-off
+pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/md.mdp b/containers/2_ApplicationSpecific/GROMACS/inputs/md.mdp
new file mode 100644
index 00000000..5b6ac2dc
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/md.mdp
@@ -0,0 +1,48 @@
+title = OPLS Lysozyme MD run
+; Run parameters
+integrator = md ; leap-frog integrator
+nsteps = 5000000 ; 2 * 2500000 = 10000 ps (10 ns)
+dt = 0.002 ; 2 fs
+; Output control
+nstxout = 0 ; suppress bulky .trr file by specifying
+nstvout = 0 ; 0 for output frequency of nstxout,
+nstfout = 0 ; nstvout, and nstfout
+nstenergy = 5000 ; save energies every 10.0 ps
+nstlog = 5000 ; update log file every 10.0 ps
+nstxout-compressed = 5000 ; save compressed coordinates every 10.0 ps
+compressed-x-grps = System ; save the whole system
+; Bond parameters
+continuation = yes ; Restarting after NPT
+constraint_algorithm = lincs ; holonomic constraints
+constraints = h-bonds ; bonds involving H are constrained
+lincs_iter = 1 ; accuracy of LINCS
+lincs_order = 4 ; also related to accuracy
+; Nonbonded settings
+cutoff-scheme = Verlet ; Buffered neighbor searching
+ns_type = grid ; search neighboring grid cells
+nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
+; vdW
+rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
+rvdw-switch = 1.0
+vdw-modifier = force-switch
+DispCorr = No
+; Electrostatics
+rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
+coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
+pme_order = 4 ; cubic interpolation
+fourierspacing = 0.16 ; grid spacing for FFT
+; Temperature coupling is on
+tcoupl = V-rescale ; modified Berendsen thermostat
+tc-grps = System
+tau_t = 1.0
+ref_t = 298
+; Pressure coupling is on
+pcoupl = C-rescale
+pcoupltype = isotropic ; uniform scaling of box vectors
+tau_p = 5.0 ; time constant, in ps
+ref_p = 1.0 ; reference pressure, in bar
+compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
+; Periodic boundary conditions
+pbc = xyz ; 3-D PBC
+; Velocity generation
+gen_vel = no ; Velocity generation is off
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/minim.mdp b/containers/2_ApplicationSpecific/GROMACS/inputs/minim.mdp
new file mode 100644
index 00000000..1af4f96c
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/minim.mdp
@@ -0,0 +1,15 @@
+; minim.mdp - used as input into grompp to generate em.tpr
+; Parameters describing what to do, when to stop and what to save
+integrator = steep ; Algorithm (steep = steepest descent minimization)
+emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
+emstep = 0.01 ; Minimization step size
+nsteps = 50000 ; Maximum number of (minimization) steps to perform
+
+; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
+nstlist = 1 ; Frequency to update the neighbor list and long range forces
+cutoff-scheme = Verlet ; Buffered neighbor searching
+ns_type = grid ; Method to determine neighbor list (simple, grid)
+coulombtype = PME ; Treatment of long range electrostatic interactions
+rcoulomb = 1.0 ; Short-range electrostatic cut-off
+rvdw = 1.0 ; Short-range Van der Waals cut-off
+pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/npt.mdp b/containers/2_ApplicationSpecific/GROMACS/inputs/npt.mdp
new file mode 100644
index 00000000..8feab0f1
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/npt.mdp
@@ -0,0 +1,47 @@
+title = OPLS Lysozyme NPT equilibration
+define = -DPOSRES ; position restrain the protein
+; Run parameters
+integrator = md ; leap-frog integrator
+nsteps = 250000 ; 2 * 250000 = 500 ps
+dt = 0.002 ; 2 fs
+; Output control
+nstxout = 500 ; save coordinates every 1.0 ps
+nstvout = 500 ; save velocities every 1.0 ps
+nstenergy = 500 ; save energies every 1.0 ps
+nstlog = 500 ; update log file every 1.0 ps
+; Bond parameters
+continuation = yes ; Restarting after NVT
+constraint_algorithm = lincs ; holonomic constraints
+constraints = h-bonds ; bonds involving H are constrained
+lincs_iter = 1 ; accuracy of LINCS
+lincs_order = 4 ; also related to accuracy
+; Nonbonded settings
+cutoff-scheme = Verlet ; Buffered neighbor searching
+ns_type = grid ; search neighboring grid cells
+nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
+; vdW
+rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
+rvdw-switch = 1.0
+vdw-modifier = force-switch
+DispCorr = No
+; Electrostatics
+rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
+coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
+pme_order = 4 ; cubic interpolation
+fourierspacing = 0.16 ; grid spacing for FFT
+; Temperature coupling is on
+tcoupl = V-rescale ; stochastic Bussi thermostat
+tc-grps = System
+tau_t = 1.0
+ref_t = 298
+; Pressure coupling is on
+pcoupl = C-rescale
+pcoupltype = isotropic ; uniform scaling of box vectors
+tau_p = 5.0 ; time constant, in ps
+ref_p = 1.0 ; reference pressure, in bar
+compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
+refcoord_scaling = com
+; Periodic boundary conditions
+pbc = xyz ; 3-D PBC
+; Velocity generation
+gen_vel = no ; Velocity generation is off
diff --git a/containers/2_ApplicationSpecific/GROMACS/inputs/nvt.mdp b/containers/2_ApplicationSpecific/GROMACS/inputs/nvt.mdp
new file mode 100644
index 00000000..432fd6a4
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/inputs/nvt.mdp
@@ -0,0 +1,44 @@
+title = OPLS Lysozyme NVT equilibration
+define = -DPOSRES ; position restrain the protein
+; Run parameters
+integrator = md ; leap-frog integrator
+nsteps = 50000 ; 2 * 50000 = 100 ps
+dt = 0.002 ; 2 fs
+; Output control
+nstxout = 2500 ; save coordinates every 5.0 ps
+nstvout = 2500 ; save velocities every 5.0 ps
+nstenergy = 2500 ; save energies every 5.0 ps
+nstlog = 2500 ; update log file every 5.0 ps
+; Bond parameters
+continuation = no ; first dynamics run
+constraint_algorithm = lincs ; holonomic constraints
+constraints = h-bonds ; bonds involving H are constrained
+lincs_iter = 1 ; accuracy of LINCS
+lincs_order = 4 ; also related to accuracy
+; Nonbonded settings
+cutoff-scheme = Verlet ; Buffered neighbor searching
+ns_type = grid ; search neighboring grid cells
+nstlist = 10 ; 20 fs, largely irrelevant with Verlet
+; vdW
+rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
+rvdw-switch = 1.0
+vdw-modifier = force-switch
+DispCorr = No ; per CHARMM FF convention
+; Electrostatics
+rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm)
+coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
+pme_order = 4 ; cubic interpolation
+fourierspacing = 0.16 ; grid spacing for FFT
+; Temperature coupling is on
+tcoupl = V-rescale ; stochastic Bussi thermostat
+tc-grps = System
+tau_t = 1.0 ; value of tau (ps)
+ref_t = 298 ; temperature (K)
+; Pressure coupling is off
+pcoupl = no ; no pressure coupling in NVT
+; Periodic boundary conditions
+pbc = xyz ; 3-D PBC
+; Velocity generation
+gen_vel = yes ; assign velocities from Maxwell distribution
+gen_temp = 298 ; temperature for Maxwell distribution
+gen_seed = -1 ; generate a random seed
diff --git a/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_long_example.bash b/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_long_example.bash
new file mode 100644
index 00000000..5cd9aa0e
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_long_example.bash
@@ -0,0 +1,423 @@
+#!/bin/bash -l
+
+## This file is intended to serve as a template to be downloaded and modified for your use case.
+## For more information, refer to the following resources whenever referenced in the script-
+## README- https://github.com/ubccr/ccr-examples/tree/main/slurm/README.md
+## DOCUMENTATION- https://docs.ccr.buffalo.edu/en/latest/hpc/jobs
+
+## Select a Slurm account that is appropriate for your use case
+## Available options and more details are provided in CCR's documentation:
+## https://docs.ccr.buffalo.edu/en/latest/hpc/jobs/#slurm-directives-partitions-qos
+#SBATCH --cluster=ub-hpc
+#SBATCH --partition=arm64
+#SBATCH --qos=arm64
+#SBATCH --account=[SlurmAccountName]
+
+#SBATCH --constraint=ARM64
+## NOTE: this line is requred to avoid odd X86_64 errors
+#SBATCH --export=HOME,TERM,SHELL
+
+## Job runtime limit, the job will be canceled once this limit is reached. Format- dd-hh:mm:ss
+#SBATCH --time=01:00:00
+
+## Refer to DOCUMENTATION for details on the following Slurm directives
+## This example uses a node with (at least) one GPU
+#SBATCH --nodes=1
+#SBATCH --tasks-per-node=1
+#SBATCH --gpus-per-node=1
+##SBATCH --cpus-per-task=28
+
+## Using conainer shared namespace when running apptainer [...] gmx mdrun [...]
+## so ask for the whole node (security measure)
+#SBATCH --exclusive
+
+## Use all the memory on the node.
+#SBATCH --mem=0
+
+
+## CONTAINER_DIR == directory with the GROMACS container image
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+
+## For the latest version of the nvidia GROMACS container see:
+## https://catalog.ngc.nvidia.com/orgs/hpc/containers/gromacs
+GROMACS_TAG="2023.2"
+
+## Set the number of OpenMPI threads per task
+## Use ${SLURM_CPUS_PER_TASK} if "#SBATCH --cpus-per-task=[...]" is used above,
+## otherwise, divide the number of CPU cores by the number of GPUs allocated to the job
+export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-$((${SLURM_JOB_CPUS_PER_NODE} / ${SLURM_GPUS_ON_NODE}))}"
+
+## Change the nvidia cache dir from ~/.nv/ComputeCache
+export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+mkdir -p "${CUDA_CACHE_PATH}"
+
+## Enable the direct GPU communication capabilities of MPI
+export GMX_ENABLE_DIRECT_GPU_COMM=1
+
+container_image="gromacs-${GROMACS_TAG}-$(arch).sif"
+
+# make sure APPTAINER_TMPDIR is set to a sensible default
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+
+## Fetch the nvidia GROMACS container, if necessary
+gromacs_url="docker://nvcr.io/hpc/gromacs:${GROMACS_TAG}"
+test ! -d "${CONTAINER_DIR}" && mkdir "${CONTAINER_DIR}"
+pushd "${CONTAINER_DIR}" > /dev/null
+if ! test -f "${container_image}"
+then
+ export APPTAINER_CACHEDIR="${APPTAINER_CACHEDIR:-${SLURMTMPDIR}/apptainer}"
+ mkdir -p "${APPTAINER_CACHEDIR}"
+ echo "Fetching the nvidia GROMACS container..."
+ apptainer --silent pull "${container_image}" "${gromacs_url}"
+ if test ! -f "${container_image}"
+ then
+ echo "apptainer pull failed - bailing!" >&2
+ exit 1
+ fi
+ echo "container downloaded sucessfully:"
+ ls -lh "${container_image}"
+ echo
+fi
+popd > /dev/null
+
+## This example uses the hen egg white lysozyme - PDB code 1AKI
+## This PDB text file was downloaded from http://www.rcsb.org/pdb/home/home.do
+if ! test -f "./inputs/1AKI.pdb"
+then
+ mkdir -p "./inputs"
+ curl -L -sS -o "./inputs/1AKI.pdb" "https://raw.githubusercontent.com/ubccr/ccr-examples/refs/heads/main/containers/2_ApplicationSpecific/GROMACS/data_files/1AKI.pdb"
+fi
+
+## Delete the crystal water molecules (residue "HOH" in the PDB file)
+grep -v "HOH" "./inputs/1AKI.pdb" > "./inputs/1AKI_clean.pdb"
+
+## Verify thtat there a no entries listed under the comment MISSING
+## Incomplete internal sequences or any amino acid residues that have missing
+## atoms will cause pdb2gmx to fail
+grep "MISSING" "./inputs/1AKI_clean.pdb"
+if [ "$?" = "0" ]
+then
+ echo "Warning!" >&2
+ echo "The \"./inputs/1AKI.pdb\" file has MISSING entries" >&2
+ echo "Incomplete internal sequences or any amino acid residues that have missing" >&2
+ echo "atoms will cause pdb2gmx to fail"
+ echo >&2
+fi
+
+## This example uses the CHARMM36 force field, downloaded from the
+## MacKerell lab website http://mackerell.umaryland.edu
+if ! test -f "charmm36-jul2022.ff.tgz"
+then
+ curl -L -sS -o "charmm36-jul2022.ff.tgz" "https://mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/charmm36-jul2022.ff.tgz"
+fi
+tar xzf "charmm36-jul2022.ff.tgz"
+
+
+## Use "gmx pdb2gmx" to generate three files:
+## The topology for the molecule.
+## A position restraint file.
+## A post-processed structure file.
+## "gmx pdb2gmx" is run as a single task:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx pdb2gmx -f ./inputs/1AKI_clean.pdb -o 1AKI_processed.gro -water tip3p -ff charmm36-jul2022
+
+## This generates three files:
+echo "output files:"
+ls -l topol.top posre.itp 1AKI_processed.gro
+echo
+
+
+## Define the box dimensions using the editconf module:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.2 -bt cubic
+
+## This generates one file
+echo "output file:"
+ls -l 1AKI_newbox.gro
+echo
+
+
+## Fill the box with solvent (water) using the solvate module
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
+
+## This generates one file, "1AKI_solv.gro" and updates topol.top
+echo "output file:"
+ls -l 1AKI_solv.gro
+echo "changes to \"topol.top\":"
+diff topol.top \#topol.top.1#
+echo
+
+
+## Download the example molecular dynamics parameter (.mdp) file from
+## http://www.mdtutorials.com/ to the "inputs" direcory
+if ! test -f "./inputs/ions.mdp"
+then
+ curl -L -sS -o "./inputs/ions.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/ions.mdp"
+fi
+
+## Generate an atomic-level input file (.tpr)
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
+
+## This generates two files:
+echo "output files:"
+ls -l ions.tpr mdout.mdp
+echo
+
+
+## Replace water molecules with the ions
+echo "SOL" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
+
+## This generates one file, "1AKI_solv_ions.gro" and updates topol.top
+echo "output file:"
+ls -l 1AKI_solv_ions.gro
+echo "changes to \"topol.top\":"
+diff topol.top '#topol.top.2#'
+echo
+
+
+## Download the input parameter file "minim.mdp" to the inputs directory
+if ! test -f "./inputs/minim.mdp"
+then
+ curl -L -sS -o "./inputs/minim.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/minim.mdp"
+fi
+
+## Run the energy minimization
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
+
+## This generates one file, "em.tpr" and updates mdout.mdp
+echo "output file:"
+ls -l em.tpr mdout.mdp
+echo
+
+
+## Run the energy minimization
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -v -deffnm em
+
+## This generates four files:
+echo "output files:"
+ls -l em.log em.trr em.edr em.gro
+echo
+
+
+## Analyze the .edr file "em.edr"
+echo "Potential" |apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f em.edr -o potential.xvg
+
+## This generates one file
+echo "output file:"
+ls -l potential.xvg
+echo
+
+
+## Equilibrate the solvent and ions around the protein:
+## Phase 1 is conducted under an NVT ensemble (constant Number of particles,
+## Volume, and Temperature.)
+
+## Download the .mdp file for this example to the inputs directory
+if ! test -f "./inputs/nvt.mdp"
+then
+ curl -L -sS -o "./inputs/nvt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/nvt.mdp"
+fi
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
+
+## This generates one file, "nvt.tpr" and updates mdout.mdp
+echo "output file:"
+ls -l nvt.tpr mdout.mdp
+echo
+
+## Run the NVT simulation
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm nvt
+
+## This generates five files:
+echo "output files:"
+ls -l nvt.cpt nvt.gro nvt.edr nvt.trr nvt.log
+echo
+
+
+## Analyze the temperature progression
+echo "Temperature" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f nvt.edr -o temperature.xvg
+
+## This generates one file
+echo "output file:"
+ls -l temperature.xvg
+echo
+
+
+## Equilibrate the solvent and ions around the protein:
+## Phase 2 - Equilibration of pressure is conducted under an NPT ensemble where
+## the Number of particles, Pressure, and Temperature are all constant
+
+# Download the 500-ps NPT equilibration .mdp file "npt.mdp" to the inputs directory
+if ! test -f "./inputs/npt.mdp"
+then
+ curl -L -sS -o "./inputs/npt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/npt.mdp"
+fi
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
+
+## This generates one file, "npt.tpr" and updates mdout.mdp
+echo "output files:"
+ls -l npt.tpr mdout.mdp
+echo
+
+
+## Run the NPT simulation
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm npt
+
+
+## Analyze the pressure progression
+echo "Pressure" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f npt.edr -o pressure.xvg
+
+## This generates one file
+echo "output file:"
+ls -l pressure.xvg
+echo
+
+
+## Examine the density using energy
+echo "Density" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f npt.edr -o density.xvg
+
+## This generates one file
+echo "output file:"
+ls -l density.xvg
+echo
+
+
+## Now the system is equilibrated, release the position restraints and run
+## production MD for data collection
+
+## Download the 10-ns MD simulation file (md.mdp) file from
+## http://www.mdtutorials.com/ to the "inputs" direcory
+if ! test -f "./inputs/md.mdp"
+then
+ curl -L -sS -o "./inputs/md.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/md.mdp"
+fi
+
+## Generate the .tpr file for this simulation:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_10.tpr
+
+## This generates one file, "md_0_10.tpr" and updates mdout.mdp
+echo "output filse:"
+ls -l md_0_10.tpr mdout.mdp
+echo
+
+
+## Run the 10-ns MD simulation:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm md_0_10
+
+## This generates six files:
+echo "output files:"
+ls -l md_0_10.log md_0_10.xtc md_0_10.edr md_0_10.gro md_0_10_prev.cpt md_0_10.cpt
+echo
+
+
+## Correcting for Periodicity Effects
+## Reimage the trajectory
+echo -e "Protein\nSystem" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_noPBC.xtc -pbc mol -center
+
+## This generates one file
+echo "output file:"
+ls -l md_0_10_noPBC.xtc
+echo
+
+
+# Root-Mean-Square Deviation
+echo -e "Backbone\nBackbone" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx rms -s md_0_10.tpr -f md_0_10_noPBC.xtc -o rmsd.xvg -tu ns
+
+## This generates one file
+echo "output file:"
+ls -l rmsd.xvg
+echo
+
+
+## Calculate RMSD relative to the crystal structure
+echo -e "Backbone\nBackbone" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx rms -s em.tpr -f md_0_10_noPBC.xtc -o rmsd_xtal.xvg -tu ns
+
+## This generates one file
+echo "output file:"
+ls -l rmsd_xtal.xvg
+echo
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_short_example.bash b/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_short_example.bash
new file mode 100644
index 00000000..b96b2814
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/slurm_ARM64_nvidia_GROMACS_short_example.bash
@@ -0,0 +1,105 @@
+#!/bin/bash -l
+
+## This file is intended to serve as a template to be downloaded and modified for your use case.
+## For more information, refer to the following resources whenever referenced in the script-
+## README- https://github.com/ubccr/ccr-examples/tree/main/slurm/README.md
+## DOCUMENTATION- https://docs.ccr.buffalo.edu/en/latest/hpc/jobs
+
+## Select a Slurm account that is appropriate for your use case
+## Available options and more details are provided in CCR's documentation:
+## https://docs.ccr.buffalo.edu/en/latest/hpc/jobs/#slurm-directives-partitions-qos
+#SBATCH --cluster=ub-hpc
+#SBATCH --partition=arm64
+#SBATCH --qos=arm64
+#SBATCH --account=[SlurmAccountName]
+
+#SBATCH --constraint=ARM64
+## NOTE: this line is requred to avoid odd X86_64 errors
+#SBATCH --export=HOME,TERM,SHELL
+
+## Job runtime limit, the job will be canceled once this limit is reached. Format- dd-hh:mm:ss
+#SBATCH --time=00:20:00
+
+## Refer to DOCUMENTATION for details on the following Slurm directives
+## This example uses a node with (at least) one GPU
+#SBATCH --nodes=1
+#SBATCH --tasks-per-node=1
+#SBATCH --gpus-per-node=1
+##SBATCH --cpus-per-task=28
+
+## Using conainer shared namespace when running apptainer [...] gmx mdrun [...]
+## so ask for the whole node (security measure)
+#SBATCH --exclusive
+
+## Use all the memory on the node.
+#SBATCH --mem=0
+
+
+## CONTAINER_DIR == directory with the GROMACS container image
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+
+## For the latest version of the nvidia GROMACS container see:
+## https://catalog.ngc.nvidia.com/orgs/hpc/containers/gromacs
+GROMACS_TAG="2023.2"
+
+## Set the number of OpenMPI threads per task
+## Use ${SLURM_CPUS_PER_TASK} if "#SBATCH --cpus-per-task=[...]" is used above,
+## otherwise, divide the number of CPU cores by the number of GPUs allocated to the job
+export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-$((${SLURM_JOB_CPUS_PER_NODE} / ${SLURM_GPUS_ON_NODE}))}"
+
+## Change the nvidia cache dir from ~/.nv/ComputeCache
+export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+mkdir -p "${CUDA_CACHE_PATH}"
+
+## Enable the direct GPU communication capabilities of MPI
+export GMX_ENABLE_DIRECT_GPU_COMM=1
+
+container_image="gromacs-${GROMACS_TAG}-$(arch).sif"
+
+# make sure APPTAINER_TMPDIR is set to a sensible default
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+
+## Fetch the nvidia GROMACS container, if necessary
+gromacs_url="docker://nvcr.io/hpc/gromacs:${GROMACS_TAG}"
+test ! -d "${CONTAINER_DIR}" && mkdir "${CONTAINER_DIR}"
+pushd "${CONTAINER_DIR}" > /dev/null
+if ! test -f "${container_image}"
+then
+ export APPTAINER_CACHEDIR="${APPTAINER_CACHEDIR:-${SLURMTMPDIR}/apptainer}"
+ mkdir -p "${APPTAINER_CACHEDIR}"
+ echo "Fetching the nvidia GROMACS container..."
+ apptainer --silent pull "${container_image}" "${gromacs_url}"
+ if test ! -f "${container_image}"
+ then
+ echo "apptainer pull failed - bailing!" >&2
+ exit 1
+ fi
+ echo "container downloaded sucessfully:"
+ ls -lh "${container_image}"
+ echo
+fi
+popd > /dev/null
+
+## Fetch the benchmarks
+if [ ! -f "water_GMX50_bare.tar.gz" ]
+then
+ wget --no-verbose http://ftp.gromacs.org/pub/benchmarks/water_GMX50_bare.tar.gz
+fi
+gzip -dc water_GMX50_bare.tar.gz | tar xf -
+
+cd water-cut1.0_GMX50_bare/0000.96/
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f pme.mdp -o bench.tpr
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -resethway -npme 0 -notunepme -noconfout -nsteps 1000 -v -s bench.tpr
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_long_example.bash b/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_long_example.bash
new file mode 100644
index 00000000..83223179
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_long_example.bash
@@ -0,0 +1,419 @@
+#!/bin/bash -l
+
+## This file is intended to serve as a template to be downloaded and modified for your use case.
+## For more information, refer to the following resources whenever referenced in the script-
+## README- https://github.com/ubccr/ccr-examples/tree/main/slurm/README.md
+## DOCUMENTATION- https://docs.ccr.buffalo.edu/en/latest/hpc/jobs
+
+## Select a cluster, partition, qos and account that is appropriate for your use case
+## Available options and more details are provided in CCR's documentation:
+## https://docs.ccr.buffalo.edu/en/latest/hpc/jobs/#slurm-directives-partitions-qos
+#SBATCH --cluster=[cluster]
+#SBATCH --partition=[partition]
+#SBATCH --qos=[qos]
+#SBATCH --account=[SlurmAccountName]
+
+## Job runtime limit, the job will be canceled once this limit is reached. Format- dd-hh:mm:ss
+#SBATCH --time=01:00:00
+
+## Refer to DOCUMENTATION for details on the following Slurm directives
+## This example uses a node with (at least) one GPU
+#SBATCH --nodes=1
+#SBATCH --tasks-per-node=1
+#SBATCH --gpus-per-node=1
+##SBATCH --cpus-per-task=28
+
+## Using conainer shared namespace when running apptainer [...] gmx mdrun [...]
+## so ask for the whole node (security measure)
+#SBATCH --exclusive
+
+## Use all the memory on the node.
+#SBATCH --mem=0
+
+
+## CONTAINER_DIR == directory with the GROMACS container image
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+
+## For the latest version of the nvidia GROMACS container see:
+## https://catalog.ngc.nvidia.com/orgs/hpc/containers/gromacs
+GROMACS_TAG="2023.2"
+
+## Set the number of OpenMPI threads per task
+## Use ${SLURM_CPUS_PER_TASK} if "#SBATCH --cpus-per-task=[...]" is used above,
+## otherwise, divide the number of CPU cores by the number of GPUs allocated to the job
+export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-$((${SLURM_JOB_CPUS_PER_NODE} / ${SLURM_GPUS_ON_NODE}))}"
+
+## Change the nvidia cache dir from ~/.nv/ComputeCache
+export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+mkdir -p "${CUDA_CACHE_PATH}"
+
+## Enable the direct GPU communication capabilities of MPI
+export GMX_ENABLE_DIRECT_GPU_COMM=1
+
+container_image="gromacs-${GROMACS_TAG}-$(arch).sif"
+
+# make sure APPTAINER_TMPDIR is set to a sensible default
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+
+## Fetch the nvidia GROMACS container, if necessary
+gromacs_url="docker://nvcr.io/hpc/gromacs:${GROMACS_TAG}"
+test ! -d "${CONTAINER_DIR}" && mkdir "${CONTAINER_DIR}"
+pushd "${CONTAINER_DIR}" > /dev/null
+if ! test -f "${container_image}"
+then
+ export APPTAINER_CACHEDIR="${APPTAINER_CACHEDIR:-${SLURMTMPDIR}/apptainer}"
+ mkdir -p "${APPTAINER_CACHEDIR}"
+ echo "Fetching the nvidia GROMACS container..."
+ apptainer --silent pull "${container_image}" "${gromacs_url}"
+ if test ! -f "${container_image}"
+ then
+ echo "apptainer pull failed - bailing!" >&2
+ exit 1
+ fi
+ echo "container downloaded sucessfully:"
+ ls -lh "${container_image}"
+ echo
+fi
+popd > /dev/null
+
+## This example uses the hen egg white lysozyme - PDB code 1AKI
+## This PDB text file was downloaded from http://www.rcsb.org/pdb/home/home.do
+if ! test -f "./inputs/1AKI.pdb"
+then
+ mkdir -p "./inputs"
+ curl -L -sS -o "./inputs/1AKI.pdb" "https://raw.githubusercontent.com/ubccr/ccr-examples/refs/heads/main/containers/2_ApplicationSpecific/GROMACS/data_files/1AKI.pdb"
+fi
+
+## Delete the crystal water molecules (residue "HOH" in the PDB file)
+grep -v "HOH" "./inputs/1AKI.pdb" > "./inputs/1AKI_clean.pdb"
+
+## Verify thtat there a no entries listed under the comment MISSING
+## Incomplete internal sequences or any amino acid residues that have missing
+## atoms will cause pdb2gmx to fail
+grep "MISSING" "./inputs/1AKI_clean.pdb"
+if [ "$?" = "0" ]
+then
+ echo "Warning!" >&2
+ echo "The \"./inputs/1AKI.pdb\" file has MISSING entries" >&2
+ echo "Incomplete internal sequences or any amino acid residues that have missing" >&2
+ echo "atoms will cause pdb2gmx to fail"
+ echo >&2
+fi
+
+## This example uses the CHARMM36 force field, downloaded from the
+## MacKerell lab website http://mackerell.umaryland.edu
+if ! test -f "charmm36-jul2022.ff.tgz"
+then
+ curl -L -sS -o "charmm36-jul2022.ff.tgz" "https://mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/charmm36-jul2022.ff.tgz"
+fi
+tar xzf "charmm36-jul2022.ff.tgz"
+
+
+## Use "gmx pdb2gmx" to generate three files:
+## The topology for the molecule.
+## A position restraint file.
+## A post-processed structure file.
+## "gmx pdb2gmx" is run as a single task:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx pdb2gmx -f ./inputs/1AKI_clean.pdb -o 1AKI_processed.gro -water tip3p -ff charmm36-jul2022
+
+## This generates three files:
+echo "output files:"
+ls -l topol.top posre.itp 1AKI_processed.gro
+echo
+
+
+## Define the box dimensions using the editconf module:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.2 -bt cubic
+
+## This generates one file
+echo "output file:"
+ls -l 1AKI_newbox.gro
+echo
+
+
+## Fill the box with solvent (water) using the solvate module
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
+
+## This generates one file, "1AKI_solv.gro" and updates topol.top
+echo "output file:"
+ls -l 1AKI_solv.gro
+echo "changes to \"topol.top\":"
+diff topol.top \#topol.top.1#
+echo
+
+
+## Download the example molecular dynamics parameter (.mdp) file from
+## http://www.mdtutorials.com/ to the "inputs" direcory
+if ! test -f "./inputs/ions.mdp"
+then
+ curl -L -sS -o "./inputs/ions.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/ions.mdp"
+fi
+
+## Generate an atomic-level input file (.tpr)
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
+
+## This generates two files:
+echo "output files:"
+ls -l ions.tpr mdout.mdp
+echo
+
+
+## Replace water molecules with the ions
+echo "SOL" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
+
+## This generates one file, "1AKI_solv_ions.gro" and updates topol.top
+echo "output file:"
+ls -l 1AKI_solv_ions.gro
+echo "changes to \"topol.top\":"
+diff topol.top '#topol.top.2#'
+echo
+
+
+## Download the input parameter file "minim.mdp" to the inputs directory
+if ! test -f "./inputs/minim.mdp"
+then
+ curl -L -sS -o "./inputs/minim.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/minim.mdp"
+fi
+
+## Run the energy minimization
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
+
+## This generates one file, "em.tpr" and updates mdout.mdp
+echo "output file:"
+ls -l em.tpr mdout.mdp
+echo
+
+
+## Run the energy minimization
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -v -deffnm em
+
+## This generates four files:
+echo "output files:"
+ls -l em.log em.trr em.edr em.gro
+echo
+
+
+## Analyze the .edr file "em.edr"
+echo "Potential" |apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f em.edr -o potential.xvg
+
+## This generates one file
+echo "output file:"
+ls -l potential.xvg
+echo
+
+
+## Equilibrate the solvent and ions around the protein:
+## Phase 1 is conducted under an NVT ensemble (constant Number of particles,
+## Volume, and Temperature.)
+
+## Download the .mdp file for this example to the inputs directory
+if ! test -f "./inputs/nvt.mdp"
+then
+ curl -L -sS -o "./inputs/nvt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/nvt.mdp"
+fi
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
+
+## This generates one file, "nvt.tpr" and updates mdout.mdp
+echo "output file:"
+ls -l nvt.tpr mdout.mdp
+echo
+
+## Run the NVT simulation
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm nvt
+
+## This generates five files:
+echo "output files:"
+ls -l nvt.cpt nvt.gro nvt.edr nvt.trr nvt.log
+echo
+
+
+## Analyze the temperature progression
+echo "Temperature" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f nvt.edr -o temperature.xvg
+
+## This generates one file
+echo "output file:"
+ls -l temperature.xvg
+echo
+
+
+## Equilibrate the solvent and ions around the protein:
+## Phase 2 - Equilibration of pressure is conducted under an NPT ensemble where
+## the Number of particles, Pressure, and Temperature are all constant
+
+# Download the 500-ps NPT equilibration .mdp file "npt.mdp" to the inputs directory
+if ! test -f "./inputs/npt.mdp"
+then
+ curl -L -sS -o "./inputs/npt.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/npt.mdp"
+fi
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
+
+## This generates one file, "npt.tpr" and updates mdout.mdp
+echo "output files:"
+ls -l npt.tpr mdout.mdp
+echo
+
+
+## Run the NPT simulation
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm npt
+
+
+## Analyze the pressure progression
+echo "Pressure" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f npt.edr -o pressure.xvg
+
+## This generates one file
+echo "output file:"
+ls -l pressure.xvg
+echo
+
+
+## Examine the density using energy
+echo "Density" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx energy -f npt.edr -o density.xvg
+
+## This generates one file
+echo "output file:"
+ls -l density.xvg
+echo
+
+
+## Now the system is equilibrated, release the position restraints and run
+## production MD for data collection
+
+## Download the 10-ns MD simulation file (md.mdp) file from
+## http://www.mdtutorials.com/ to the "inputs" direcory
+if ! test -f "./inputs/md.mdp"
+then
+ curl -L -sS -o "./inputs/md.mdp" "http://www.mdtutorials.com/gmx/lysozyme/Files/md.mdp"
+fi
+
+## Generate the .tpr file for this simulation:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f inputs/md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_10.tpr
+
+## This generates one file, "md_0_10.tpr" and updates mdout.mdp
+echo "output filse:"
+ls -l md_0_10.tpr mdout.mdp
+echo
+
+
+## Run the 10-ns MD simulation:
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -deffnm md_0_10
+
+## This generates six files:
+echo "output files:"
+ls -l md_0_10.log md_0_10.xtc md_0_10.edr md_0_10.gro md_0_10_prev.cpt md_0_10.cpt
+echo
+
+
+## Correcting for Periodicity Effects
+## Reimage the trajectory
+echo -e "Protein\nSystem" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_noPBC.xtc -pbc mol -center
+
+## This generates one file
+echo "output file:"
+ls -l md_0_10_noPBC.xtc
+echo
+
+
+# Root-Mean-Square Deviation
+echo -e "Backbone\nBackbone" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx rms -s md_0_10.tpr -f md_0_10_noPBC.xtc -o rmsd.xvg -tu ns
+
+## This generates one file
+echo "output file:"
+ls -l rmsd.xvg
+echo
+
+
+## Calculate RMSD relative to the crystal structure
+echo -e "Backbone\nBackbone" | apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx rms -s em.tpr -f md_0_10_noPBC.xtc -o rmsd_xtal.xvg -tu ns
+
+## This generates one file
+echo "output file:"
+ls -l rmsd_xtal.xvg
+echo
+
diff --git a/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_short_example.bash b/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_short_example.bash
new file mode 100644
index 00000000..bcf843d3
--- /dev/null
+++ b/containers/2_ApplicationSpecific/GROMACS/slurm_nvidia_GROMACS_short_example.bash
@@ -0,0 +1,101 @@
+#!/bin/bash -l
+
+## This file is intended to serve as a template to be downloaded and modified for your use case.
+## For more information, refer to the following resources whenever referenced in the script-
+## README- https://github.com/ubccr/ccr-examples/tree/main/slurm/README.md
+## DOCUMENTATION- https://docs.ccr.buffalo.edu/en/latest/hpc/jobs
+
+## Select a cluster, partition, qos and account that is appropriate for your use case
+## Available options and more details are provided in CCR's documentation:
+## https://docs.ccr.buffalo.edu/en/latest/hpc/jobs/#slurm-directives-partitions-qos
+#SBATCH --cluster=[cluster]
+#SBATCH --partition=[partition]
+#SBATCH --qos=[qos]
+#SBATCH --account=[SlurmAccountName]
+
+## Job runtime limit, the job will be canceled once this limit is reached. Format- dd-hh:mm:ss
+#SBATCH --time=00:20:00
+
+## Refer to DOCUMENTATION for details on the following Slurm directives
+## This example uses a node with (at least) one GPU
+#SBATCH --nodes=1
+#SBATCH --tasks-per-node=1
+#SBATCH --gpus-per-node=1
+##SBATCH --cpus-per-task=28
+
+## Using conainer shared namespace when running apptainer [...] gmx mdrun [...]
+## so ask for the whole node (security measure)
+#SBATCH --exclusive
+
+## Use all the memory on the node.
+#SBATCH --mem=0
+
+
+## CONTAINER_DIR == directory with the GROMACS container image
+CONTAINER_DIR="/projects/academic/[CCRgroupname]/Containers"
+
+## For the latest version of the nvidia GROMACS container see:
+## https://catalog.ngc.nvidia.com/orgs/hpc/containers/gromacs
+GROMACS_TAG="2023.2"
+
+## Set the number of OpenMPI threads per task
+## Use ${SLURM_CPUS_PER_TASK} if "#SBATCH --cpus-per-task=[...]" is used above,
+## otherwise, divide the number of CPU cores by the number of GPUs allocated to the job
+export OMP_NUM_THREADS="${SLURM_CPUS_PER_TASK:-$((${SLURM_JOB_CPUS_PER_NODE} / ${SLURM_GPUS_ON_NODE}))}"
+
+## Change the nvidia cache dir from ~/.nv/ComputeCache
+export CUDA_CACHE_PATH="${SLURMTMPDIR:-/var/tmp}/nv_$(id -nu)"
+mkdir -p "${CUDA_CACHE_PATH}"
+
+## Enable the direct GPU communication capabilities of MPI
+export GMX_ENABLE_DIRECT_GPU_COMM=1
+
+container_image="gromacs-${GROMACS_TAG}-$(arch).sif"
+
+# make sure APPTAINER_TMPDIR is set to a sensible default
+export APPTAINER_TMPDIR="${APPTAINER_TMPDIR:-${SLURMTMPDIR}/apptainer/tmp}"
+mkdir -p "${APPTAINER_TMPDIR}"
+
+## Fetch the nvidia GROMACS container, if necessary
+gromacs_url="docker://nvcr.io/hpc/gromacs:${GROMACS_TAG}"
+test ! -d "${CONTAINER_DIR}" && mkdir "${CONTAINER_DIR}"
+pushd "${CONTAINER_DIR}" > /dev/null
+if ! test -f "${container_image}"
+then
+ export APPTAINER_CACHEDIR="${APPTAINER_CACHEDIR:-${SLURMTMPDIR}/apptainer}"
+ mkdir -p "${APPTAINER_CACHEDIR}"
+ echo "Fetching the nvidia GROMACS container..."
+ apptainer --silent pull "${container_image}" "${gromacs_url}"
+ if test ! -f "${container_image}"
+ then
+ echo "apptainer pull failed - bailing!" >&2
+ exit 1
+ fi
+ echo "container downloaded sucessfully:"
+ ls -lh "${container_image}"
+ echo
+fi
+popd > /dev/null
+
+## Fetch the benchmarks
+if [ ! -f "water_GMX50_bare.tar.gz" ]
+then
+ wget --no-verbose http://ftp.gromacs.org/pub/benchmarks/water_GMX50_bare.tar.gz
+fi
+gzip -dc water_GMX50_bare.tar.gz | tar xf -
+
+cd water-cut1.0_GMX50_bare/0000.96/
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx grompp -f pme.mdp -o bench.tpr
+
+apptainer run \
+ -B /projects:/projects,/scratch:/scratch,/util:/util,/vscratch:/vscratch \
+ --sharens \
+ --nv \
+ "${CONTAINER_DIR}/${container_image}" \
+ gmx mdrun -ntmpi ${SLURM_GPUS_ON_NODE} -resethway -npme 0 -notunepme -noconfout -nsteps 1000 -v -s bench.tpr
+